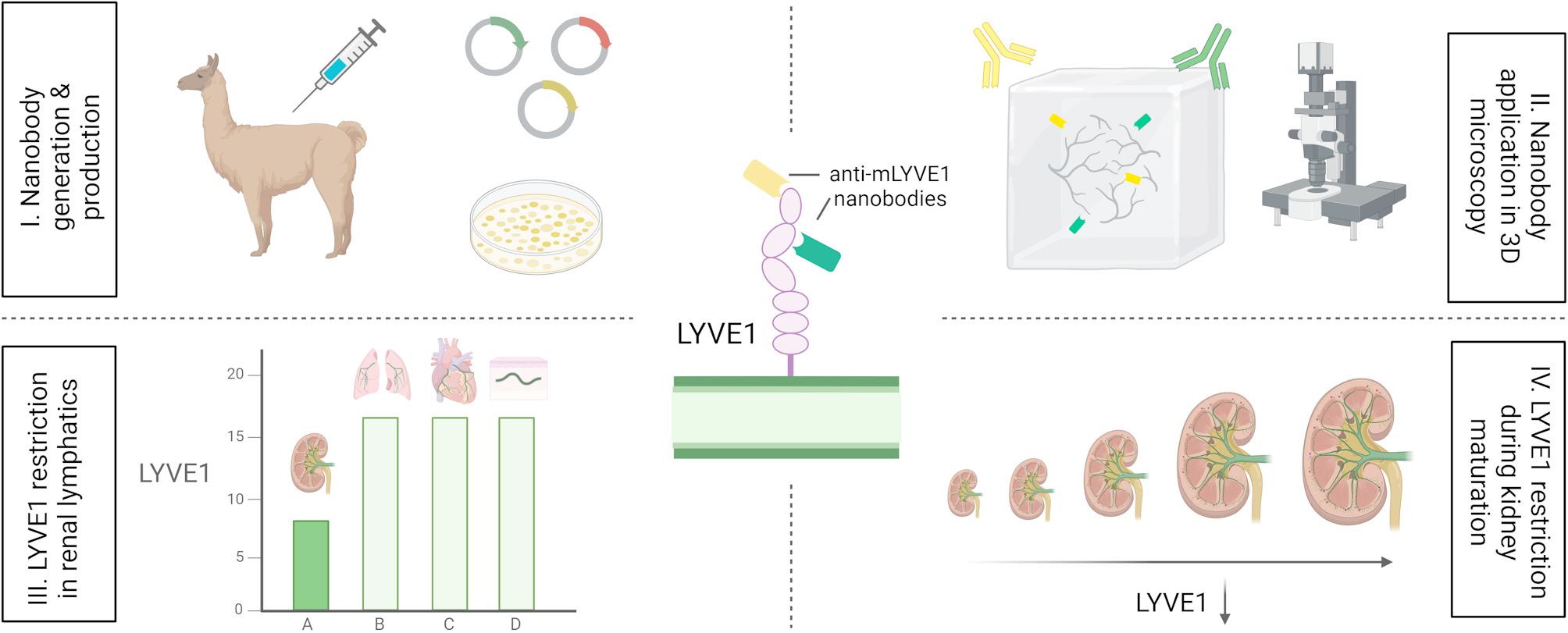
Llama immunization and nanobody library construction
For nanobody generation, a recombinant murine LYVE1 protein, consisting of 288 amino acids, fused to a His10 tag, was expressed in human embryonic kidney 293 cells. The recombinant protein was injected alongside Adjuvant P (3111, GERBU Biotechnik GmbH, Heidelberg, Germany) weekly on six consecutive occasions into a Ilama (Lama glama). Forty days after the initial immunization, 100 mL of anticoagulated blood were collected from the Ilama, 5 days after the final antigen injection. Peripheral blood lymphocytes were isolated and total RNA extracted for cDNA first-strand synthesis using oligo(dT) primers. Sequences encoding variable nanobodies were subsequently amplified by PCR, digested with the restriction enzyme SapI, and cloned into the SapI site of the phagemid vector pMECS. Electrocompetent E. coli TG1 cells (60502, Lucigen, Middleton, WI, USA) were then transformed with the variable domain of heavy-chain-only antibody (VHH) sequence harbouring pMECS vectors, resulting in a nanobody library comprising 109 independent transformants. This process has previously been described in detail [34].
Biopanning and identification of anti-mouse nanobodies specific to LYVE1
Phage enrichment and biopanning were carried out as described [34]. Briefly, the previously constructed nanobody library was panned for 3 rounds on solid phase coated with mouse LYVE1 (100 µg/mL in 100 mM NaHCO3, pH 8.2) yielding a 400-fold enrichment of antigen-specific phages after the 3rd round of panning. A total of 380 colonies were randomly selected and assayed for mouse LYVE1-specific antigens by ELISA, again using mouse LYVE1 and additionally mouse LYVE1 fused to human IgG1 Fc at the C-terminus (50065-M02H, Sino Biological, Beijing, China). To exclude potential nanobody clones binding to human IgG1 Fc, human IgG1 Fc (10702-HNAH, Sino Biological) was utilized as a control, as well as blocking buffer only (100 mM NaHCO3, pH 8.2). After comparing binding specificity of different clones with the control values, 278 colonies were identified as positive for mouse LYVE1 binding. Using the sequence data, the number of possible nanobody candidates was further narrowed down to 98, of which 96 were able to specifically bind both mouse LYVE1-His10 and mouse LYVE1 fused to human IgG1 Fc. The remaining unique clones derived from 21 different B cell lineages according to their complementary determining region (CDR) 3 groups. Considering the different B cell linages and robustness of ELISA screening data, 6 clones were selected for further experiments.
Cloning of nanobody sequences in expression vector
To generate 6xHis-tagged nanobodies, nanobody sequences were cloned from the pMECS phagemid vector into the pHEN6c expression vector. Initially, nanobody sequences were amplified by PCR using the following primers:
(1) 5’ GAT GTG CAG CTG CAG GAG TCT GGR GGA GG 3’.
(2) 5’ CTA GTG CGG CCG CTG AGG AGA CGG TGA CCT GGG T 3’.
PCR products were purified (28104, QIAquick PCR Purification Kit,, Qiagen, Hilden, Germany) and digested for 20 min at 37 °C with PstI-HF (R3140, New England Biolabs, Ipswich, MA, USA) and BstEII-HF (R3162, New England Biolabs) restriction enzymes, while empty pHEN6c plasmids were concurrently digested with the same restriction enzymes. The empty pHEN6c plasmids, however, were supplemented by 5 units of heat-inactivated (5 min at 80 °C) FastAP™ alkaline phosphatase (EF0651, Thermo Fisher Scientific, Waltham, MA, USA). Digestion products were purified (28104, QIAquick PCR Purification Kit, Qiagen) and subjected to T4 DNA ligase-mediated ligation reactions. The ligation reaction was performed at 16 °C for 16 h using 2.5 units of T4 DNA ligase (M0202, New England Biolabs). Subsequently, newly generated nanobody sequence harbouring pHEN6c plasmids were transformed into WK6 E. coli cells (C303006, Thermo Fisher Scientific) and analysed to determine correct nanobody sequence integration by Sanger DNA sequencing using the following primers:
(1) 5’ TCA CAC AGG AAA CAG CTA TGA C 3’.
(2) 5’ CGC CAG GGT TTT CCC AGT CAC GAC 3’.
Production and purification of anti-LYVE1 nanobodies
WK6 E. coli carrying pHEN6c-Nanobody plasmid were cultivated at 37 °C, shaking in 1 L `Terrific Broth` medium (2.3 g/L KH2PO4, 16.4 g/L K2HPO4-3H2O, 12 g/L tryptone, 24 g/L yeast extract, 0.4% (v/v) glycerol) complemented with 100 µg/mL ampicillin, 2 mM MgCl2, and 0.1% (w/v) glucose. Nanobody expression was induced at an OD600 of 0.6–0.9 by adding 1 nM isopropyl ß-D-1-thiogalactopyranoside (IPTG). After an incubation period of 16 h, the nanobodies were extracted by centrifugation (8000× g, 8 min, RT). 18 mL of TES/4 buffer (0.05 M Tris [pH 8.0], 0.125 mM EDTA, 0.125 M sucrose) were added for 1 h of shaking on ice. The cell suspension was then centrifuged (8000× g, 30 min, 4 °C), and periplasmic protein-containing supernatant collected. For 6xHis-tagged nanobody extraction, HIS-Select® nickel affinity gel (P6611, Sigma-Aldrich, Darmstadt, Germany) was applied according to the manufacturer’s instructions. The solution was loaded on a PD-10 column (17-0435-01, GE healthcare, Chicago, IL, USA) and nanobodies eluted via 3 × 1 mL 0.5 M imidazole in phosphate-buffered saline (PBS) (I2399, Sigma-Aldrich). An overnight dialysis (3 kDa MWCO, 66382, Thermo Fisher Scientific) against PBS was carried out to remove undesirable imidazole from the nanobody solution.
Coomassie-blue stained SDS PAGE and Western blotting
To verify nanobody production and pureness, sample protein was separated by molecular weight using established sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). For each clone, 5 µg of denatured protein with 0.04% (w/v) OrangeG in ddH2O) were loaded onto the gel alongside a pre-stained protein ladder (ab116029, Abcam, Cambridge, UK). For protein visualisation, gels were treated with Coomassie staining solution (0.1% (w/v), Coomassie Brilliant Blue R-250 (1610400, Bio-Rad Laboratories Inc., Hercules, CA, USA), 50% (v/v) methanol, and 10% (v/v) glacial acetic acid in ddH2O for 1 h, followed by incubation with Coomassie destaining solution (50% ddH2O, 40% methanol, 10% acetic acid (v/v/v)). Alternatively, gels were blotted onto a nitrocellulose membrane (1620112, Bio-Rad Laboratories Inc.) and nanobodies identified using a primary anti-His antibody (12698, Cell Signaling Technology, Danvers, MA, USA) and a secondary anti-rabbit antibody (926-32211, LI-COR Biosciences, Lincoln, NE, USA). Subsequently, blots were analysed by an Odyssey® Fc Imaging System (LI-COR Biosciences).
Mouse husbandry and acquisition of mouse tissues
C57BL/6 wildtype mice, or Lyve1Cre − eGFP mice [35] on the C57BL/6 background were utilised in this study. All animal experiments were carried out under a UK Home Office project license (PPL: PP1776587) in compliance with the UK Animals (Scientific Procedures) Act 1986 or approved by German federal authorities (LaGeSo Berlin) under the licence number ZH120. Nutrition and water were available to animals ad libitum. Mouse embryos were staged by time-matings, considering embryonic day (E) 0.5 to be the morning a copulation plug was detected. Adult mice or pregnant mice were sacrificed using CO2 inhalation and cervical translocation as a Schedule 1 procedure. The desired organs were obtained from embryonic, juvenile, or adult mice, washed in PBS, and subsequently fixed in 4% (w/v) paraformaldehyde (PFA) in PBS for 4 h at 4 °C to preserve tissue integrity. After fixation, samples were thoroughly washed in three changes of PBS and stored at 4 °C in PBS containing 0.03% (w/v) sodium azide until further processing.
Antibodies for Immunofluorescence
The following commercially available antibodies were used: rabbit monoclonal anti-His antibody (12698, Cell Signaling Technologies) [1:200], donkey polyclonal anti-rabbit IgG Alexa Fluor™ 647 antibody (A31573, Invitrogen, Waltham, MA, USA) [1:1000], donkey polyclonal anti-rabbit IgG Highly-Cross-Absorbed Alexa Fluor™ 647 antibody (A32795, Invitrogen) [1:1000], goat polyclonal anti-mLYVE1 (AF2125, R&D Systems, Minneapolis, MN, USA) [1:100], donkey polyclonal anti-goat IgG Alexa Fluor™ 568 antibody (A11057, Invitrogen) [1:1000], donkey polyclonal anti-goat IgG Highly cross-absorbed Alexa Fluor™ 488 + antibody (A32814, Invitrogen) [1:1000], hamster monoclonal anti-Podoplanin (14-5381-82, Invitrogen) [1:200], goat polyclonal anti-Syrian hamster IgG Cross-Absorbed Alexa Fluor™ 546 antibody (A-21111, Invitrogen) [1:1000], chicken polyclonal anti-GFP (ab13970, Abcam) [1:200], donkey anti-chicken Highly cross-absorbed Alexa Fluor™ 488 + antibody (A32931TR, Invitrogen) [1:1000], rat monoclonal anti-mF4/80 antibody (MCA497G, BioRad) [1:50], donkey anti-rat Highly cross-absorbed Alexa Fluor™ 488 + antibody (A48269, Invitrogen) [1:1000]. Nanobodies were detected by anti-His staining in combination with an Alexa Fluor™ dye-conjugated secondary antibody. An overview of antibody combinations used for each experiment including concentration and incubation time can be found in Table S1.
Zenon labelling of IgG antibodies
Anti-His antibodies were labelled using the Zenon™ Rabbit IgG Labeling Kit (Z25306, Thermo Fisher Scientific) by incubating the antibody with Component A for 5 min, followed by blocking with Component B for another 5 min. The labelled antibody was used within 30 min.
Direct labelling of nanobodies
Anti-mouse LYVE1 nanobodies were directly labelled with Alexa Fluor™ 647 NHS-Ester (A20006, Invitrogen) by reacting 1 mg of nanobody with 1 mg of dye in NaHCO₃ buffer (pH 8.0) for 1 h at room temperature. Labelled nanobodies were purified using a NAP™-10 column (Sephadex™ G-25, Thermo Fisher) and stored in PBS with 0.03% (w/v) sodium azide.
Immunofluorescence staining of cryosections
Snap-frozen 5 μm sections of E14.5 wildtype mice were stained with nanobodies (0.1 µg/mL) and appropriate control antibodies as previously described [36]. Visualisation of representative regions was accomplished using an Axioscope5 fluorescence microscope (Zeiss, Oberkochen, Germany) equipped with a Plan-NEOFLUAR 40x/0.75 objective (Zeiss).
Wholemount Immunofluorescence staining and optical clearing
Mouse organs were either cut into smaller sections measuring 0.5–2 mm thickness (for studies in Figs. 2A, 3 and 4, Fig. S3B) using a scalpel at room temperature or processed as whole specimens (for studies in Figs. 2B and 4A (E18.5-P5), Fig. S3A, Fig. S4). Tissues were dehydrated using an ascending methanol series and bleached overnight at 4 °C in a solution containing 5% H2O2 (VWR Chemicals, Radnor, PE, USA) in methanol. Wholemount staining was performed as previously described [22]. For optical clearing, samples were treated with methanol for dehydration followed by BABB (1:2 benzyl alcohol and benzyl benzoate) immersion, as described in [22]. Intestine samples were not optically-cleared, but mounted in fluorescent medium (S3023, Agilent Technologies, Santa Clara, CA, USA). Details on antibodies used, incubation periods and concentrations are outlined in Table S1.
Wholemount Immunofluorescence staining and optical clearing of embryonic and postnatal day one specimens
Embryonic and early postnatal (P) kidneys were initially dehydrated and bleached as described above. Following rehydration, samples were permeabilized overnight in a 5% solution of 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate in ddH2O and blocked in PBS supplemented with 0.2% (v/v) Triton X100, 10% (v/v) DMSO, and 6% (v/v) goat serum. Nanobodies (10 µg/mL) and antibodies were diluted in antibody solution (PBS + 0.2% v/v Tween20 + 0.1% v/v heparin solution + 5% v/v DMSO + 3% v/v goat serum + 0.1% w/v saponin) and incubated for a duration between 4 (nanobodies) and 24 h (IgG antibodies) at 4 °C. Between staining steps, samples were washed in PBS-Tween20. Clearing was performed with BABB as previously described for embryonic kidneys [37].
Confocal and light sheet imaging
Specimens were imaged using two different microscopy systems. For confocal imaging, an LSM 880 Upright Confocal Multiphoton microscope (Zeiss) equipped with a 20x/NA 1.0 W-plan Apochromat water immersion objective was utilized. A comprehensive description of the setup specific to BABB-cleared specimens for this microscope can be found in previously published work [37]. Intact mouse organs were imaged by LaVision Ultramicroscope II with a LaVison BioTec MVPLAPO 2x OC OBE objective. Various magnifications were employed, and image acquisition utilized a step size of 2 μm. Imaging thresholds were set individually for each channel during image acquisition and post-processing. This approach was chosen to accommodate the variability in autofluorescence and other factors, and to ensure optimal signal detection for each channel.
Image processing and 3D rendering
2D immunofluorescence images were subjected to post-processing using ZEN 3.4 (blue edition) software from Zeiss. Single channels were extracted and saved in TIFF format. Z-stack datasets were processed and 3D-rendered using Imaris (version 9.8, Oxford Instruments Abingdon, UK). To reduce non-specific background signal, single channels were subjected to the Isosurface Render function. Images of the 3D-rendered data were captured using the Snapshot function and saved in TIFF format. Videos were captured using the Animation function.
Quantification of signal-to-background ratio
Signal-to-background ratio was quantified using ImageJ 2.24/1.54f (https://github.com/imagej/ImageJ [38]), . Ten intensity values were randomly selected in vessel areas and areas that showed no specific staining. The mean values for signal and background areas were calculated and the signal-to-background ratio was determined using the formula: Signal-to-background ratio = mean signal/mean background.
Calculation of correlation coefficients
Pearson’s correlation coefficients r were calculated using the Imaris Coloc function. For very large microscopy files, colocalization analysis was performed on representative regions of interest, as full-volume analysis was not computationally feasible on the available hardware. For colocalization of nuclear GFP staining and endothelial nanobody staining, an object-based approach was applied. Spots were segmented independently for GFP and LYVE1 signals using the Imaris Spots function. For each LYVE1 spot, the shortest distance to the nearest GFP spot was determined. LYVE1 spots within 25 μm of a GFP spot were classified as colocalized, and the coefficient was calculated as the proportion of colocalized LYVE1 spots relative to the total number of LYVE1 spots.
Quantitative analysis of 3D imaging volumes
The quantitative analysis of three-dimensional volumes commenced with the binarization of single channels using Imaris. The binarization process was conducted using the Isosurface Render function, and for E18.5 and P1 samples, 3D cropping was applied to exclude regions with high podoplanin (PDPN) intensity at the kidney surface, thus enhancing binarization accuracy. During surface rendering, the threshold was set to absolute intensity, with manual adjustments to ensure the inclusion of all relevant structures. To eliminate smaller non-specific signals, structures were filtered based on the number of voxels. Following surface generation, PDPN channel-derived non-lymphatic structures, such as glomeruli, were manually removed using the selection function. Subsequently, the channel of interest was masked using specific settings: constant inside/outside, setting voxels outside the surface to 0.00, and inside the surface to the maximum intensity of the prepared channel. All channels except the newly created masked channel were then deleted, and the single channel was saved in TIFF format. With the binarized files prepared, TIFF files were imported into the VesselVio application [39]. The analysis settings were configured as follows: unit µm, resolution type anisotropic with individual sizes of samples, analysis dimensions 3D, image resolution 1.0 µm3, and filters applied to isolate segments shorter than 10.0 µm and purge end-point segments shorter than 10.0 µm. Analysis results were automatically saved in Microsoft Excel files by VesselVio. Among the parameters offered by VesselVio, vessel volume was selected as the parameter for further analysis, as it considers both vessel length and width. To account for variability in sample size and volume, vessel volumes (LYVE1 and PDPN) were normalized by dividing the measured vessel volume by the total sample volume, calculated based on the Imaris output for the x, y, and z dimensions. This normalized value was referred to as ‘relative volume’ [40].
Single-cell RNA sequencing analysis
The generation of the mouse kidney scRNA-seq data used in this study has been described [41]. In brief, a regional enrichment strategy was used to isolate hilum, cortex and medulla separately from 12-week-old C57Bl/6 mouse kidneys (n = 14), before single-cell droplet capture using the 10X Genomics platform (10x Genomics, Pleasanton, USA) and sequencing using an Illumina HiSeq X system (San Diego, USA). The count matrix corresponding to the lymphatic cluster was isolated and re-processed, applying a standard workflow using Seurat (v5) in R [42]. The Harmony package [43] was used for integration, using the mouse identifier as a batch variable. Differential expression analysis was performed using the FindAllMarkers function, utilising Wilcoxon rank sum tests. Marker genes are presented with average log-fold change (log2FC) values and an adjusted p value of < 0.05 was considered as statistically significant. Gene ontology (GO) analysis was applied using the Panther database [44], using statistical overrepresentation tests and calculating the false discovery rate (FDR) for each GO term.
Sample size Estimation and statistical analysis
Sample size was estimated based on prior publications of developmental studies of kidney lymphatic vessels [40] and studies investigating lymphatics in adult mice organs [9, 16]. These indicated that 6–8 animals per experiment would be sufficient to power statistical analyses. Statistical analysis was conducted using Prism (v8, GraphPad by Dotmatics, Boston, MA, USA) and RStudio version 12.0 (Posit, Boston, MA, USA). To determine the significance of differences in volume between LYVE1 and PDPN at individual time points and individual organs, a paired student’s t-test was carried out. For comparisons of Pearson correlation coefficients r, one-way ANOVA followed by Tukey’s post hoc test was applied. To assess statistical significance across different time points a rank-based approach using the R package nparcomp [45] with the function mctp1 (multiple comparisons for relative contrast effect testing) was chosen. This approach allows for nonparametric multiple comparisons to evaluate relative contrast effects. A p value of less than 0.05 was considered statistically significant for all tests. Quantitative data was visualised using Prism. All graphical representations present individual data points either by region of interest or by animal, along with the mean and standard error of the mean.
Preparation of figures and videos
The graphical abstract was created in BioRender.com. The figures presented in this paper were prepared using the free software tool Inkscape 1.1.0 (Inkscape Project, 2020) for graphic design and layout. Any modifications made to the images, such as adjustment of brightness and contrast, were applied consistently throughout one panel to maintain uniformity across all coherent single and merged channel images. Videos were edited in Clipchamp (Microsoft, Redmond, WA, USA).