
vivo today announced the official launch of the vivo Y21d, the latest addition to its Y Series lineup. Designed for young consumers and everyday users, the Y21d delivers long-lasting performance, dependable durability, and practical…
Blog
-
6 terrorists killed, 7 policemen martyred in DI Khan training school attack – samaa tv
- 6 terrorists killed, 7 policemen martyred in DI Khan training school attack samaa tv
- Security forces repulse attack on DI Khan police training centre; at least 3 terrorists killed Dawn
- 7 Cops, 6 Terrorists Dead In Suicide Attack At Pak Police…
Continue Reading
-

Northern Ireland v Germany: Conor Bradley will be big loss for Michael O’Neill’s side at Windsor Park
Now, the side face the prospect of taking on the group favourites without their best player.
Bradley, who has also been booked three times in five Premier League appearances this term, was shown his second yellow card of the qualifying campaign in…
Continue Reading
-

Radiation-induced miR-208a Predicts Cancer Recurrence and Death Risk i
Introduction
Existing data shows that the incidence of lung cancer has been significantly increased for few decades globally.1,2 Especially in developed countries, authoritative statistical data from China reported that lung cancer has become…
Continue Reading
-

Do You Know How Expensive Gold Is In Pakistan? 10 Grams Costs…
While India and Pakistan may differ on many fronts, one passion unites both nations: a love for gold. From weddings to festivals, gold plays a central role in celebrations. Yet, the price Pakistanis pay for gold far exceeds what is paid in India, reaching levels that are hard to imagine.

Recent reports indicate that the price of 10 grams of 24-carat gold in Pakistan is 430,500 Pakistani rupees. At such rates, purchasing gold would be prohibitively expensive for many.

The Pakistani rupee is significantly weaker than the Indian rupee. Even after conversion, Pakistanis pay roughly Rs 13,000 more than Indians for every 10 grams of gold, making it considerably more expensive there.

This naturally raises a business idea: “If we buy gold in India at lower prices and sell it in Pakistan, we could earn lakhs of rupees in profit.” The price difference makes such a venture seem highly lucrative.

Though the idea sounds lucrative, it isn’t that simple. Exporting gold from India requires government permission, and taking it across the border without authorization is illegal and a serious criminal offence.

The situation in Pakistan is different. To stabilize their economy, the government often imposes bans on gold imports. Recently, a 60-day ban led to a shortage, driving gold prices sharply higher.

Thinking about smuggling gold would be a grave mistake. Illegally transporting gold is smuggling; if caught, the metal will be confiscated, the money lost, and one could face jail time, risking one’s reputation and livelihood.

The low value of the Pakistani rupee is the main reason for this price difference, making it a relief that gold remains more affordable in our country.
Continue Reading
-

Goldman Sachs Declares Preferred Stock Dividends
NEW YORK, October 10, 2025 – The Goldman Sachs Group, Inc. (NYSE: GS) today announced that it has declared dividends on the following series of its non-cumulative preferred stock (represented by depositary shares, each representing a 1/1,000th interest in a share of preferred stock, except for the Series O Preferred Stock, the Series T Preferred Stock, the Series V Preferred Stock, the Series X Preferred Stock, and the Series Y Preferred Stock in which each depositary share represents a 1/25th interest in a share of preferred stock):
- $330.99 per share of Floating Rate Non-Cumulative Preferred Stock, Series A;
- $330.99 per share of Floating Rate Non-Cumulative Preferred Stock, Series C;
- $325.93 per share of Floating Rate Non-Cumulative Preferred Stock, Series D;
- $662.50 per share of 5.30% Fixed-to-Floating Rate Non-Cumulative Preferred Stock, Series O;
- $475.00 per share of 3.80% Fixed-Rate Reset Non-Cumulative Preferred Stock, Series T;
- $515.63 per share of 4.125% Fixed-Rate Reset Non-Cumulative Preferred Stock, Series V;
- $937.50 per share of 7.50% Fixed-Rate Reset Non-Cumulative Preferred Stock, Series X; and
- $765.63 per share of 6.125% Fixed-Rate Reset Non-Cumulative Preferred Stock, Series Y.
In addition, The Goldman Sachs Group, Inc. has declared a dividend of $1,300.04 per share of Perpetual Non-Cumulative Preferred Stock, Series E, and a dividend of $1,300.67 per share of Perpetual Non-Cumulative Preferred Stock, Series F.
The dividends on the Series A Preferred Stock, Series C Preferred Stock, Series D Preferred Stock, Series O Preferred Stock, Series T Preferred Stock, Series V Preferred Stock, Series X Preferred Stock, and Series Y Preferred Stock are payable on November 10, 2025, to preferred shareholders of record on October 26, 2025. The dividends on the Series E Preferred Stock and Series F Preferred Stock are payable on December 1, 2025, to preferred shareholders of record on November 16, 2025.
###
Goldman Sachs is a leading global financial institution that delivers a broad range of financial services to a large and diversified client base that includes corporations, financial institutions, governments and individuals. Founded in 1869, the firm is headquartered in New York and maintains offices in all major financial centers around the world.
Media Contact:
Tony Fratto
Tel: +1 212 902 5400Investor Contact:
Jehan Ilahi
Tel: +1 212 902 0300Continue Reading
-

Cristiano Ronaldo’s staff working conditions speak volumes as net worth soars to £1billion
The working conditions at Cristiano Ronaldo’s hotels have been laid bare as he basks in becoming the world’s first billionaire footballer
New billionaire
Continue Reading
-
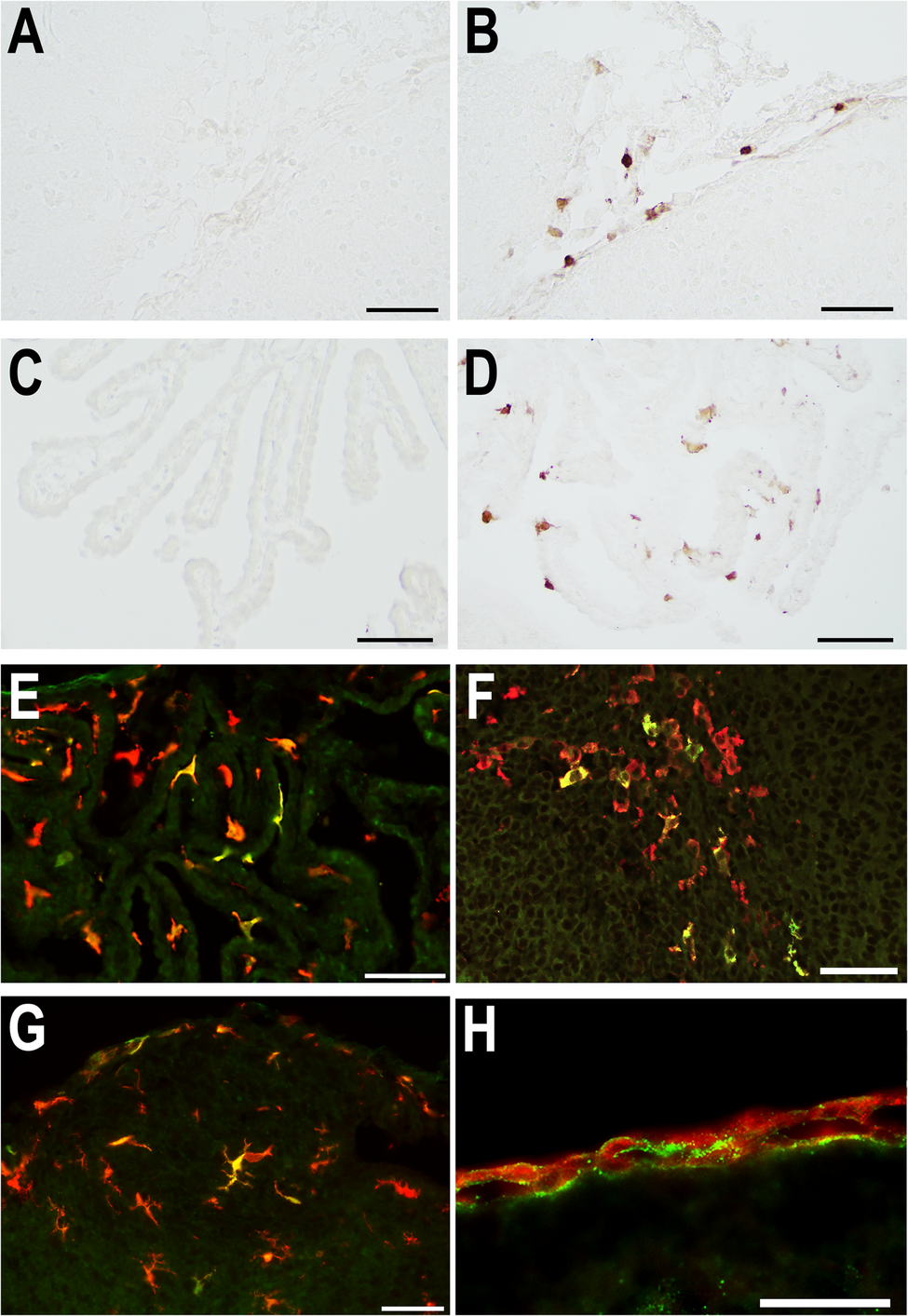
Early immune responses to systemic inflammation in the postnatal mouse brain initiated by migrating macrophages and leptomeningeal fibroblasts | Journal of Neuroinflammation
The Guide for the Care and Use of Laboratory Animals, 8th edition (National Research Council Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Washington DC: National Academies Press, 2011), was followed for the handling of all mice. The Institutional Animal Care and Use Committee of the Kyorin University Faculty of Health Sciences approved all of the experiments described (Protocols I17–08–03 to I17–08–07).
Induction of systemic inflammation in mouse pups
Systemic inflammation was induced in P7, male, C57BL/6 N mice using LPS (from E. coli O55:B5; Sigma-Aldrich-Merck, Burlington, MA, USA), a bacterial endotoxin, which was administered intraperitoneally to the experimental group at a dose of 0.75 mg/kg with a 33-G Hamilton syringe (systemic inflammation group). The control group received a single intraperitoneal injection of saline at the same dose of 3.75 mL/kg as the experimental group (saline control group). After treatment, the mouse pups were returned to their dams for continued rearing.
Preparation of histological frozen brain sections
At 4, 12, 24, 48, and 72 h after LPS or saline administration, ketamine-xylazine anesthetic solution was administered intraperitoneally at a dose of 10 mL/kg. Blood was extracted transcardially using a phosphate-buffered saline (PBS) solution followed by perfusion with Zamboni fixative solution at a flow rate of 3 mL/min. Following removal of the scalp, eyes, and mandible, the skulls containing the brain, the liver, and the spleen were immersed in Zamboni fixative at 4 °C for 2 days. Each experimental group consisted of four mice.
The brains were then extracted from the skulls. Each brain was bisected along the parasagittal plane located in the midline interhemispheric fissure so that the first parasagittal brain section was safely cut from the right hemisphere. Small fragments of the liver and spleen were prepared and embedded in the same blocks as the bisected brains. Cryoprotection was achieved through immersion of the tissue samples in 10, 15, and 20% sucrose in PBS at room temperature overnight. Brain, liver, and spleen tissues were then embedded in Cryomatrix embedding medium (Thermo Fisher Scientific, Waltham, MA, USA) in Tissue Tech Cryomold No. 3 (Sakura Finetech, Tokyo, Japan) and subsequently frozen with dry ice-cold n-hexane.
Frozen blocks were sectioned at a thickness of 14 μm using a LEICA CM 3050 S cryostat (Leica Biosystems, Deer Park, IL, USA). Median brain sections were obtained by cutting sequentially from 300 μm to the right of the interhemispheric fissure of the cerebral hemispheres. Approximately 40 sections were prepared as median sections. Lateral brain sections were prepared from 600 μm to the left of the interhemispheric fissure and sequentially cut to obtain 50 lateral sections. The prepared sections were mounted on FRC-04 coated glass slides (Matsunami Glass Co., Ltd., Osaka, Japan), followed by air drying and vacuum drying using a V-100 vacuum pump (BUCHI Labortechnik, Flawil, Switzerland). The dried sections were then stored at −20 °C until use.
Sections from mice in all experimental groups were stained with hematoxylin and eosin (H&E) to observe inflammation-related changes, such as inflammatory cell infiltration, ischemia, and necrosis. The sections were subsequently used for immunohistochemical, double immunofluorescence, and in situ hybridization (ISH) staining. To examine the histology of major organs other than the brain by H&E staining, the heart, lung, liver, spleen, and kidney were embedded in paraffin and sectioned at a thickness of 4 μm using a microtome.
Immunohistochemistry and Immunofluorescence staining
Frozen sections were soaked in Tris-buffered saline with Tween 20 (TBS-T) for 10 min and pretreated with 0.3% H2O2 in methanol to block endogenous peroxidase activity and with 1% bovine serum albumin (BSA) in TBS-T to block non-specific binding. Sections were incubated with primary antibodies (Table 1) overnight at 4 °C or for 2 h at room temperature, followed by incubation with reagents from the ImmPRESS Horseradish Peroxidase (HRP) Antibody Polymer Detection Kit (Vector Laboratories, Burlingame, CA, USA) for 60 min at room temperature. Reactions were visualized by incubating sections with an ImmPACT 3,3’-Diaminobenzidine (DAB) Substrate Kit, Peroxidase (SK-4105; Vector Laboratories). Sections were sequentially dehydrated through 80%, 90%, 95%, and 100% ethanol, cleared with xylene, and coverslipped using a mounting medium (HSR, Sysmex, Kobe, Japan). Immunohistochemical photographs were taken with 4x, 10x, 20x, and 40x PlanApo λ objectives (Nikon, Tokyo, Japan) of an Eclipse Ci-L light microscope equipped with a DS-Fi3/DS-L4 digital camera control unit (Nikon), and with 4x, 10x, 20x, and 40x PlanApo λ objectives (Nikon) of a BZ-X710 microscope (Keyence, Osaka, Japan).
Table 1 Primary antibodies for immunohistochemical and immunofluorescence staining For double immunofluorescence staining, frozen sections were soaked in TBS-T for 10 min, preincubated with 1% BSA in TBS-T, and incubated with primary antibodies overnight at 4 °C. The combinations of the primary antibodies were IL-1β and ionized calcium-binding adaptor molecule-1 (Iba1), CCL2 and type 1 collagen, or IL-1R1 and type 1 collagen. After incubation with two primary antibodies, sections were incubated with donkey anti-goat or anti-rabbit IgG secondary antibodies conjugated with Alexa Fluor 568 or 488 (Invitrogen-Thermo Fisher Scientific, Waltham, MA, USA) for 60 min at room temperature. Nuclear counterstaining was performed with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. Sections were coverslipped with Fluorescence Mounting Medium (DAKO, Agilent, Santa Clara, CA, USA). Fluorescence images were captured using a BZ-X710 microscope equipped with structured illumination.
Morphometric analysis of sections immunohistochemically stained with anti-Iba1
To evaluate the changes of microglial morphology that occurred after LPS administration, frozen brain sections from mice at 4, 24, 48, and 72 h after LPS or saline administration (n = 4 in each group) were immunohistochemically stained with anti-Iba1 antibody (rabbit monoclonal [EPR16588], Abcam, Cambridge, UK). For morphometric studies, a computerized image analyzer (WinROOF 2018, Mitani Corporation, Tokyo, Japan) was used to calculate the area fraction of Iba1-positive cells (total area of Iba1-positive cell bodies with cytoplasmic processes divided by the area of interest). In addition to the analysis functions, WinROOF 2018 had a variety of combined manual and automatic editing functions, such as separating contiguous cytoplasmic process images, filling in parts of cells with density below the threshold, and removing small cell fragments and artifacts. Each field was reviewed by the operator on the instrument screen to make these corrections. Analysis of the hippocampus, cerebral cortex, and cerebellum was performed. The number of cell bodies of Iba1-positive cells was counted in the hippocampus and cerebral cortex. Two histological sections per individual mouse were used for each brain region.
For the hippocampus, the area was delineated from the lateral parasagittal sections located between 700 and 1100 μm lateral to the interhemispheric fissure. The hippocampus was not divided into subregions for quantification of the Iba1-positive area of each analyzed section (Supplementary Fig. 1). In the same sections in which the hippocampus was analyzed, the parietal cortex was located dorsal to the hippocampus, and the six cortical layers were perfectly recognizable. To define the field of view for the analysis of the cerebral cortex, the first line was drawn from the point on the cortico-medullary junction corresponding to the caudal end of the lateral ventricle (point a, Supplementary Fig. 1) and perpendicular to the cortical surface (point b). The second line was drawn from point c, which was located 500 μm caudally from point b on the cortical surface, perpendicular to the cortico-medullary junction (point d). The arachnoid and pia on the cortical surface were also included in the area of interest. In the cerebellum, median sections were used for morphometric analysis. Notably, the cerebellum did not include the deep cerebellar nuclei, and the entire cerebellar section consisted of the cerebellar cortex and medulla and the leptomeninges (Supplementary Fig. 1).
Immunoassay of cytokine concentrations in brain parenchymal tissues
At 4, 24, 48, and 72 h after LPS or saline administration, mice were perfused systemically with PBS to remove blood. The brains were removed and quickly divided on an ice-cold glass plate into the following seven regions: left and right cerebral cortices, left and right limbic systems (including olfactory bulb, olfactory tubercle, piriform cortex, entorhinal cortex, and hippocampus), left and right subcortical structures (striatum, diencephalon, midbrain, and brainstem), and cerebellum. These seven brain parts were individually snap frozen in liquid nitrogen and stored at −80 °C until they were used.
Of the seven parts, the left cerebral cortex, left limbic system, and cerebellum were used to measure cytokine concentrations. Tissue Protein Extraction Reagent (T-PER, Thermo Fisher Scientific) was added to a Biomasher II tube (Nippi, Tokyo, Japan) at 20-fold tissue weight, and 1/100 volume of Halt Protease Inhibitor Cocktail (100x, Thermo Fisher Scientific) was added to T-PER. Tissue samples stored at −80 °C were added to these tubes and homogenized. After centrifugation at 13,000 rpm, 4 °C, for 5 min, only the supernatant was collected and used as the protein extraction solution. The extract was dispensed into 50-µL portions into microtubes and stored at −80 °C. Protein yield was measured by colorimetric quantification using bicinchoninic acid (BCA Protein Assay Kit, TaKaRa, Shiga, Japan) and serial dilution of BSA. Using the Luminex 200 xPONENT system (Thermo Fisher Scientific), a simultaneous multiplex protein immunoassay system of protein extracts from the left cerebral cortex, left limbic system, and cerebellum was prepared to determine the tissue concentrations of the following 15 cytokines: CCL2, CCL11, CXCL1, CXCL2, CXCL10, G-CSF, IL-1α, IL-1β, IL-4, IL-6, IL-10, IL-12, IL-17, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α).
Isolation of CD11b(+) cells, RNA extraction, and microarray analysis
At 24 h after LPS or saline administration, mice were deeply anesthetized with ketamine and xylazine. Blood was washed out by transcardial perfusion with sterile Dulbecco’s phosphate-buffered saline [D-PBS(−)] to remove plasma and blood cells. Fresh whole brains, including the arachnoid, pia, and parenchyma, were quickly removed. Two brains from littermates that were treated in the same experimental manner were placed in a 50-mL conical tube containing ice-cold D-PBS(−) and processed as a single sample. Four samples were prepared in each of the systemic inflammation and control groups.
To collect macrophages and microglial cells from fresh brains, CD11b(+) cells were isolated using magnetic-activated cell sorting (MACS) methods [28]. Whole brains were dissociated by enzymatic digestion of the extracellular matrix using the Adult Brain Dissociation Kit for mice (Miltenyi Biotec, Auburn, CA, USA). Mechanical dissociation steps were performed using the gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec) according to the manufacturer’s protocol. Brain tissue dissociates were centrifuged at 400 × g for 5 min at 4 °C. The pellets were resuspended in cold D-PBS (+) (with calcium and magnesium) containing 0.5% BSA. In the presence of the kit’s Debris Removal Solution, the cell suspensions were centrifuged at 3000 × g for 12 min at 4 °C to remove the debris phase. The pellets were resuspended in cold D-PBS (+) containing 0.5% BSA and incubated with R-phycoerythrin (PE)-conjugated primary human/mouse CD11b monoclonal antibody (130–113–235, Miltenyi Biotec) and Fc receptor blocking reagent (130–092–575, Miltenyi Biotec), followed by incubation with MicroBeads UltraPure conjugated to anti-PE monoclonal antibody (130–105–639, Miltenyi Biotec). Suspended cells labeled with anti-PE MicroBeads were enriched by magnetic separation using an LS column (Miltenyi Biotec) that was placed in a QuadroMACS separator (Miltenyi Biotec) according to the manufacturer’s protocol. CD11b-positive-selected cells (positive fraction) were considered macrophages and microglia. During the positive selection process, CD11b-negative cells were also collected (negative fraction). The numbers of cells in the positive and negative fractions were determined using cell counting plates (OneCell counter; Fine Plus International, Kyoto, Japan) under an ECLIPSE Ts2 (Nikon) inverted phase-contrast microscope. The cells of the positive and negative fractions were finally suspended in 1 mL CELLBANKER 1 Plus (TaKaRa) and stored at − 80 °C before RNA extraction.
Frozen cells were thawed rapidly at 37 °C, centrifuged at 400 × g for 5 min at 4 °C, and washed with RNase-free PBS by centrifugation under the same conditions. Total RNA was extracted from the cell pellets using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. RNA quantification was performed using NanoVue (GE Healthcare Life Sciences, Chicago, IL, USA) and 2100 BioAnalyzer (Agilent, Santa Clara, CA, USA). Eight RNA samples were of high quality with RNA Integrity Numbers (RINs) ranging from 7.9 to 9.1.
Gene expression profiles were analyzed by Hokkaido System Science Co., Ltd. (Hokkaido, Japan) using the SurePrint G3 Mouse Gene Expression (GE) 8 × 60 K Ver.2.0 Microarray (Agilent, G4852B). Cyanine-3 (Cy3)-labeled cRNA was prepared from 50 ng of total RNA using the Low Input Quick Amp Labeling Kit (Agilent), followed by RNeasy column purification (QIAGEN). Then, 0.6 µg of Cy3-labeled cRNA (specific activity > 6 pmol Cy3/µg cRNA) was fragmented at 60 °C for 30 min in a reaction volume of 25 µL containing 25x Fragmentation Buffer (Agilent) and 10x GE Blocking Agent (Agilent). On completion of the fragmentation, 25 µL of 2x Hi-RPM GE Hybridization Buffer (Agilent) were added. The fragmentation mixture was hybridized to SurePrint G3 Mouse GE 8 × 60 K Ver.2.0 Microarray at 65 °C for 17 h in an Agilent rotating hybridization oven. After hybridization, the microarrays were washed with GE Wash Buffer 1 (Agilent) for 1 min at room temperature and with GE Wash Buffer 2 (Agilent) for 1 min at 37 °C. The slides were scanned immediately after washing on the SureScan Microarray Scanner (G2600D, Agilent) using one color scan setting for 8 × 60 K array slides (dye channel set to green, and green photomultiplier tube set to 100%). The scanned images were analyzed with Feature Extraction Software 12.0.3.1 (Agilent) using default parameters to obtain background-subtracted and spatially detrended processed signal intensities. The 75th percentile shift normalization was performed using GeneSpring GX 14.9 (Agilent), and baseline transformation was performed using the median of all samples.
Differentially expressed genes were identified using GeneSpring GX 14.9. Quality control was performed by filtering genes based on the “Detected” flag; only genes detected in at least one of the two groups were included in the analysis. Statistical analysis was conducted using an unpaired t-test with asymptotic p-value computation, followed by Benjamini-Hochberg correction for multiple testing. Genes with a corrected p-value less than 0.05 and an absolute fold change of 2.0 or greater were considered differentially expressed.
Real-time reverse transcription-polymerase chain reaction (RT-qPCR)
At 4, 12, 24, 48, and 72 h after LPS or saline administration (n = 4 samples in each group), mice were deeply anesthetized with ketamine-xylazine, blood was poured out, and fresh whole brains were quickly removed. The brains of two P7 littermate mice, which were treated in the same experimental manner, were pooled in ice-cold D-PBS(−) and processed as a single sample. CD11b(+) cells were isolated by MACS, and total RNA was extracted from CD11b(+) and CD11b(−) cells. RNA quantification was performed using NanoVue (GE Healthcare Life Sciences) and using 4150 TapeStation System (Agilent). Forty RNA samples were of high quality with RINs ranging from 8.9 to 9.8.
Fifty nanograms of total RNA were used for reverse transcription to cDNA using SuperScript III Reverse Transcriptase (Invitrogen-Thermo Fisher Scientific). The real-time reverse transcription-polymerase chain reaction (RT-qPCR) was performed using TaqMan Fast Advanced Master Mix (Applied Biosystems-Thermo Fisher Scientific), TaqMan primer/probe sets for 10 targets (Applied Biosystems-Thermo Fisher Scientific), and a 7500 Fast Real-Time PCR System (Applied Biosystems-Thermo Fisher Scientific) according to the manufacturer’s protocols. The targets were as follows: (1) Saa3 (encoding serum amyloid A3), Mm00441203_m1; (2) Saa1 (encoding serum amyloid A1), Mm00656927_g1; (3) Saa2 (encoding serum amyloid A2), Mm04208126_mH; (4) Irg1 (encoding immune-responsive gene 1 [IRG1], also known as aconitate decarboxylase 1 [ACOD1]), Mm01224532_m1; (5) Ccl5 (encoding chemokine CCL5), Mm01302428_m1; (6) Cxcl13 (encoding chemokine CXCL13), Mm00444534_m1; (7) Slfn4 (encoding schlafen-4), Mm01298330_m1; (8) Cxcl9 (encoding chemokine CXCL9), Mm00434946_m1; (9) Lcn2 (encoding lipocalin-2), Mm01324470_m1; and (10) internal control, Hprt (encoding hypoxanthine phosphoribosyltransferase), Mm03024075_m1. Analysis of relative transcript levels was performed using the ΔΔCT method. All assays were performed in triplicate.
In situ hybridization (ISH)
ISH was performed to determine which cells upregulated gene expression using the histological brain sections. Frozen sections were prepared from mice at 4, 12, 24, and 48 h after LPS or saline administration (n = 4 mice in each group) and used for ISH. ISH was performed with RNAscope 2.5 HD Assay-Brown for fixed frozen tissue (#322310; Advanced Cell Diagnostics, Newark, CA, USA) according to the manufacturer’s protocol with minor modification. The modification was as follows: Protease Plus was diluted 1:2 and incubated for 10 min, and DAB precipitation was performed using the ImmPACT DAB Substrate Kit, Peroxidase. Target probes for the RNAscope manual assay (Advanced Cell Diagnostics) were Mm-Saa3 (Cat. No. 446841), Mm-Irg1 (450241), Mm-Cxcl13 (406311), Mm-Ccl5 (469601), Mm-Cxcl9 (489341), Mm-Lcn2 (313971), Mm-Slfn4 (573011), and Mm-Col1a1 (319371).
RNAscope DAB precipitation was coupled to immunohistochemistry using anti-Iba1 antibody (rabbit monoclonal [EPR16588], Abcam) or anti-type I collagen antibody (rabbit monoclonal [EPR24331-53], Abcam). The ImmPRESS-Alkaline Phosphatase (AP) Horse Anti-Rabbit IgG Polymer Detection Kit, Alkaline Phosphatase (MP-5401, Vector), was used as the secondary antibody. Immunohistochemistry was visualized using the 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) Substrate Kit, AP (SK-5400, Vector). Sections were covered with G-Mount (Genostaff, Tokyo, Japan) and then coverslipped using a mounting medium (HSR, Sysmex). Mouse brain areas were identified with the help of anatomical atlases [29,30,31].
Statistical analysis
For the tissue cytokine immunoassay and the morphometry of Iba1-immunopositive cells, data were obtained from 8 experimental conditions (saline vs. LPS administration; 4, 24, 48, and 72 h after administration), and the results were analyzed by two-way analysis of variance (ANOVA; main effects of treatment and time). For RT-qPCR analyses, the mean ΔCT values for each gene target were obtained from 10 experimental conditions (saline vs. LPS administration; 4, 12, 24, 48, and 72 h after administration). The results were analyzed by two-way ANOVA. Post hoc tests were performed using Tukey’s test. P values less than 0.05 were considered significant in all analyses.
Continue Reading
-
Ultra-Fast IPS Monitors – Trend Hunter
- Ultra-Fast IPS Monitors Trend Hunter
- Philips Announces the Evnia 25M2N3200U A High-Performance eSports Monitor TechPowerUp
- Philips’ new 310Hz gaming monitor is perfect for esports without breaking the bank PC Guide
- Philips Launches Evnia…
Continue Reading
-
Advancing Understanding and Treatment of Cancer Cachexia: Future Chall
Introduction
Cancer cachexia (CC) is a severe metabolic disorder commonly observed in advanced cancer patients, characterized by persistent loss of skeletal muscle mass and unresponsive weight loss to conventional nutritional interventions.1,2…
Continue Reading