WASHINGTON, D.C. (7News) — Researchers at George Washington University are studying artificial intelligence to see if the technology could help people quit smoking.
Smoking is a hard habit to kick. Less than one in 10 adults who smoke…

WASHINGTON, D.C. (7News) — Researchers at George Washington University are studying artificial intelligence to see if the technology could help people quit smoking.
Smoking is a hard habit to kick. Less than one in 10 adults who smoke…

The final piece of China’s cross-border data transfer framework has now been released with the issuance of the Certification Measures. Effective January 1, 2026, businesses must closely monitor certification institutions, standards, and application procedures. Early preparation and strategic planning will be essential for long-term compliance and risk management.
On October 14, 2025, the Cyberspace Administration of China (CAC) and the State Administration for Market Regulation (SAMR) jointly issued the long-awaited Measures for Certification of Cross-Border Personal Information Transfer (hereinafter referred to as the “Measures”), which will officially take effect on January 1, 2026.
The release of the Measures marks a pivotal moment in China’s data governance landscape. It completes the three-pathway framework for cross-border personal information transfers established under the Personal Information Protection Law (PIPL).
Find Business Support
With the certification method now fully defined, China’s regulatory architecture for cross-border data transfer (CBDT) is considered comprehensive and operational.
The Measures clarify key aspects of the certification process, including scope and applicability, application procedures, certification body obligations, as well as supervision and enforcement.
This final regulatory piece enhances legal certainty for businesses. It offers enterprises another structured compliance pathway and may prove especially beneficial for multinational corporations engaged in frequent or large-scale data transfers.
Under Article 38 of PIPL, personal information processors in China who need to transfer personal data overseas for business or operational purposes must choose one of three legally prescribed pathways:
Since 2022, China has gradually built out this framework through a series of regulatory instruments. The Security Assessment Measures, released in July 2022, laid out detailed procedures for high-risk data transfers. In February 2023, the Standard Contract Measures were issued and came into effect in June, providing a more accessible compliance route for many businesses.
Find Business Support
However, the certification pathway remained incomplete for some time. While several technical standards and guidelines were released – such as the Security Certification Specifications for Cross-Border Processing of Personal Information V2.0 (December 2022) and the Information Security Technology – Certification Requirements for Cross-Border Transmission of Personal Information (March 2023) – a formal regulatory document was still missing.
Now with the release of the Measures, this long-awaited document provides the legal and procedural foundation for certification, aligning with earlier standards and the CAC’s January 2025 draft for public consultation.
Moreover, China’s State Administration for Market Regulation (SAMR) and Standardization Administration of China (SAC) jointly released the Data security technology—Security certification requirements for cross-border processing activity of personal information (GB/T 46068-2025), which will take effect on March 1, 2026.
All these developments signal that the three pathways for CBDT under the PIPL are now fully operational, marking a significant milestone in China’s data governance regime.
The newly released Measures for Certification of Cross-Border Personal Information Transfer outline the specific conditions under which personal information processors may opt for the certification pathway to legally transfer personal data overseas.
To be eligible for certification, a personal information processor must meet all of the following criteria:
Importantly, the Measures prohibit data volume splitting or other circumvention tactics to avoid the security assessment requirement. If the data transfer volume exceeds the thresholds for certification, the processor must undergo a security assessment instead.
Before applying for certification, personal information processors must fulfill several legal obligations, including:
The PIPIA must evaluate:
The PIPIA report should be retained for at least three years.
Also read: How to Conduct a Personal Information Protection Impact Assessment in China
Processors must apply for certification through a professional certification institution authorized to conduct personal information protection audits. For overseas processors, the application must be submitted via a designated domestic representative or entity.
Once the application is approved, the institution will issue a certificate, valid for three years. To maintain continuity, processors must reapply six months before the certificate expires.


The Measures establish a multi-layered oversight mechanism:
Under China’s CBDT mechanisms, both the standard contract and certification pathways provide legal mechanisms for cross-border transfers of personal information. While they share many similarities, such as overlapping applicability and similar pre-transfer obligations, their structural differences make them suitable for distinct business scenarios.
The standard contract is a self-managed process in which enterprises sign a fixed-format agreement with the overseas recipient, strictly following the CAC’s template. After conducting a self-assessment, the enterprise submits the contract and related materials for filing with the provincial CAC, which may conduct formal or substantive reviews. As a commercial agreement, the standard contract is not publicly disclosed, and its contents are not subject to public scrutiny or external evaluation.
In contrast, certification is conducted by third-party professional institutions based on CAC-issued rules. It involves a comprehensive review of the enterprise’s technical, organizational, and governance measures. Unlike the standard contract, certification carries a degree of public authority – it reflects, to some extent, administrative recognition of the enterprise’s data protection capabilities. For companies with high reputational stakes in personal information protection, certification offers a credible external endorsement of their compliance posture.
Find Business Support
The compliance focus also differs. The standard contract emphasizes the legal obligations of a specific data transfer, ensuring the overseas recipient agrees to uphold data subject rights and assumes clear data protection responsibilities. Certification, on the other hand, assesses the enterprise’s overall compliance framework, including internal governance, data protection systems, and technical safeguards. It promotes ongoing compliance and dynamic supervision. In practice, the standard contract is more suited to “one-off” or occasional transfers, while certification is better aligned with enterprises engaged in frequent or long-term cross-border data activities.
Post-transfer supervision further distinguishes the two. With the standard contract, enterprises are responsible for monitoring the overseas recipient’s compliance, and CAC oversight is primarily conducted through the filing system. Certification bodies, however, implement continuous monitoring mechanisms. Certificates may be suspended or revoked, and violations are publicly disclosed, creating external compliance pressure.
Given these differences, the standard contract is generally more appropriate for low-volume, low-risk transfers. It is relatively easy to implement but offers limited flexibility due to its fixed format. Certification, by contrast, is better suited for enterprises with frequent, high-risk, or high-profile data transfers, or those seeking to demonstrate a high level of data protection. The certification is valid for three years, helping reduce repetitive compliance efforts.
Enterprises should assess their business scale, data export scenarios, and compliance capacity to select the most appropriate pathway for CBDT.
| Aspect | Standard Contract | Certification |
| Legal nature and review mechanism | Enterprises sign a fixed-format agreement with the overseas recipient, following CAC’s template.
Self-assessment and filing with provincial CAC; subject to formal or substantive review. |
Conducted by third-party institutions under CAC rules. Reviews technical, organizational, and governance measures. Carries independent credibility. |
| Compliance focus and depth | Focuses on contractual obligations. Ensures overseas recipient upholds data subject rights and assumes data protection responsibilities. | Evaluates full compliance framework, including governance, systems, and safeguards.
Emphasizes ongoing and dynamic compliance. |
| Post-transfer supervision and accountability | Enterprises monitor recipients’ compliance. CAC oversight via the filing system. | Certification bodies provide continuous monitoring (e.g., annual inspection). Certificates may be suspended or revoked; violations are publicly disclosed. |
| Applicability and flexibility | Best for low-volume, low-risk transfers. Easy to implement, but limited flexibility due to fixed format. | Suited for frequent or high-risk transfers. Valid for three years, reducing repetitive compliance. |
| Cost | Self-declaration will not incur any cost
|
The certification body will charge relevant fees.
|
As the Measures prepare to take effect on January 1, 2026, enterprises should go beyond understanding the basic provisions and actively monitor several key developments to ensure compliant and efficient implementation.
Key areas to monitor include:
Companies are suggested to consult the official websites of the CAC, SAMR, and the Certification and Accreditation Administration of China (CNCA) for timely updates and implementation guidance.
Personal information protection certification is a critical mechanism for enabling compliant, trustworthy, and internationally aligned data flows. As the Measures enter into force in 2026, enterprises must pay close attention to the qualifications of certification institutions, evolving standards and procedures, and long-term compliance obligations. For legal, compliance, and information security professionals, now is the time to study regulatory trends, plan ahead, and strengthen internal capabilities to ensure a smooth transition into the new regime.
If you need further interpretation or practical support, our team at Dezan Shira & Associates is here to assist you with tailored guidance and hands-on expertise. For more information, please get in touch with China@dezshira.com.

Disgraced Lostprophets singer Ian Watkins died after being stabbed in the neck in a alleged prison attack, an inquest has heard.
Watkins, 48, died on 11 October after being assaulted at HMP Wakefield, where he had been serving a 29-year sentence…

Deep inside a Swiss mountain, a group of students spent some of the summer simulating what life might be like inside a lunar base. The BBC joined them before the “mission”.
What was your childhood dream? For some, it was the idea of becoming an…

Prince Andrew was paid tens of thousands of pounds by a British businessman linked to a wealth management company which ripped off pension savers.
The King’s brother, who last week said he would no longer use his titles including the Duke of York, has long faced questions about his finances and how he is funding his lifestyle.
He stepped down as a working royal in 2019 because of his association with the sex offender Jeffrey Epstein and no longer receives any money from the King.
Although his finances remain opaque, details of some of his arrangements and controversial business associates have occasionally emerged from court cases.
Prince Andrew did not respond to requests for comment.
Documents from the High Court in London show that Andrew received £60,500 from a British businessman, Adrian Gleave in December 2019, a few weeks after the BBC Newsnight interview which led to his withdrawal from public life.
The payments came to light in a High Court case brought by an elderly Turkish millionaire, Nebahat Isbilen, who claimed money she had paid to Andrew and his ex-wife Sarah Ferguson had been misappropriated by a business adviser.
That money was funnelled through a British company owned by Mr Gleave, Alphabet Capital Limited.
According to an “agreed statement of facts signed by or on behalf of the Duke and Duchess, Mr Gleave and Alphabet”, Mr Gleave’s company Alphabet had “previously made, and might in the future make, substantial payments to HRH Prince Andrew the Duke of York”.
The payments Prince Andrew received directly from Mr Gleave and his businesses, which were also sent via Alphabet Capital, came months after the businessman had stepped down as a director of SVS Securities – a company which had been ordered to stop trading by the financial regulator over pension mis-selling allegations.
SVS Securities collapsed in August 2019, days after the Financial Conduct Authority (FCA) had ordered it to stop regulated activities.
Clients’ pension funds were found to have been invested in high-risk bonds against their interests in order to generate large commissions for SVS.
Some investments made on the basis of the undisclosed commissions then defaulted, leaving customers with substantial losses. Investors were also charged large fees to withdraw funds in an effort to boost profits, the FCA found.
Mr Gleave, 55, was head of business development at SVS, which he had joined in 2013.
He remained a registered director on the FCA’s register until late July 2019, less than two weeks before the regulator’s intervention.
He had been a company director registered with Companies House until a couple of months earlier, although on LinkedIn he claims to have left the business in November 2018.
Three SVS directors were later banned and fined by the FCA but Mr Gleave was not one of them. Two are appealing the decision.
The Financial Services Compensation Scheme has paid out more than £41m to former SVS customers.
At the time of his dealings with Prince Andrew, Mr Gleave also ran a number of caravan and mobile home parks in Northern Ireland and England.
At one point, he was reported to have worked out of one of the parks, a retirement village for over-55s on the east coast of Northern Ireland.
Ten of the parks have since gone into administration and Mr Gleave, who did not respond to a request for comment, now works for a renewable energy company with a focus on AI and crypto financing.
Neither Prince Andrew nor Mr Gleave have ever explained the reason for the payments or the nature of any contractual relationship between the two men.
Baroness Margaret Hodge, a former chair of the Commons public accounts committee, said Mr Gleave’s business background raised questions for Prince Andrew about his judgement and financial dealings.
“This is yet another instance where a dose of transparency would help answer legitimate questions about the origins of the money and the purpose of the payment,” she said.
“Without those answers any sceptical person would be worried that there might be some financial wrongdoing taking place and this would risk sullying the reputation of the Royal family,” she added.
As well as the payments he made himself, Mr Gleave’s company, Alphabet Capital, was also used to funnel significant sums which had originated from Ms Isbilen to Prince Andrew and his ex-wife Sarah, court documents show.
Ms Ferguson was paid £50,000 by Alphabet Capital in February 2020. It has previously been reported that she was paid £20,000 by Alphabet for a role advising the company and that she also received more than £200,000 to cover work she had done as a brand ambassador for a US solar energy company.
Prince Andrew was separately given £750,000 directly by Ms Isbilen, money which he has repaid.
Another £10,000 was paid from Alphabet Capital to the couple’s daughter, Eugenie. This, along with a £15,000 payment from Ms Isbilen’s business adviser has previously been described by Eugenie as a gift from a “long-standing family friend” which she said was to pay for a surprise birthday party for her mother, Sarah.
Alphabet Capital filed accounts claiming it was a dormant company at the time of the payments. These were later corrected but listed a turnover of just £80,000.
Prince Andrew and Mr Gleave did not respond to requests for comment.

ITV’s hit drama Trigger Point, starring Vicky McClure (Line of Duty, Alex Rider), as Explosives Officer, or Expo, Lana Washington, an Afghan War veteran, is returning to U.K. screens for season 3 on Sunday. And it is set to be another…

Selena Gomez proved that some…

Red-ball cricket has long been considered the true test of a player’s skill, temperament, and character. For Pakistan, a nation with a proud yet turbulent cricketing history, the future of its Test setup depends heavily on how well it…
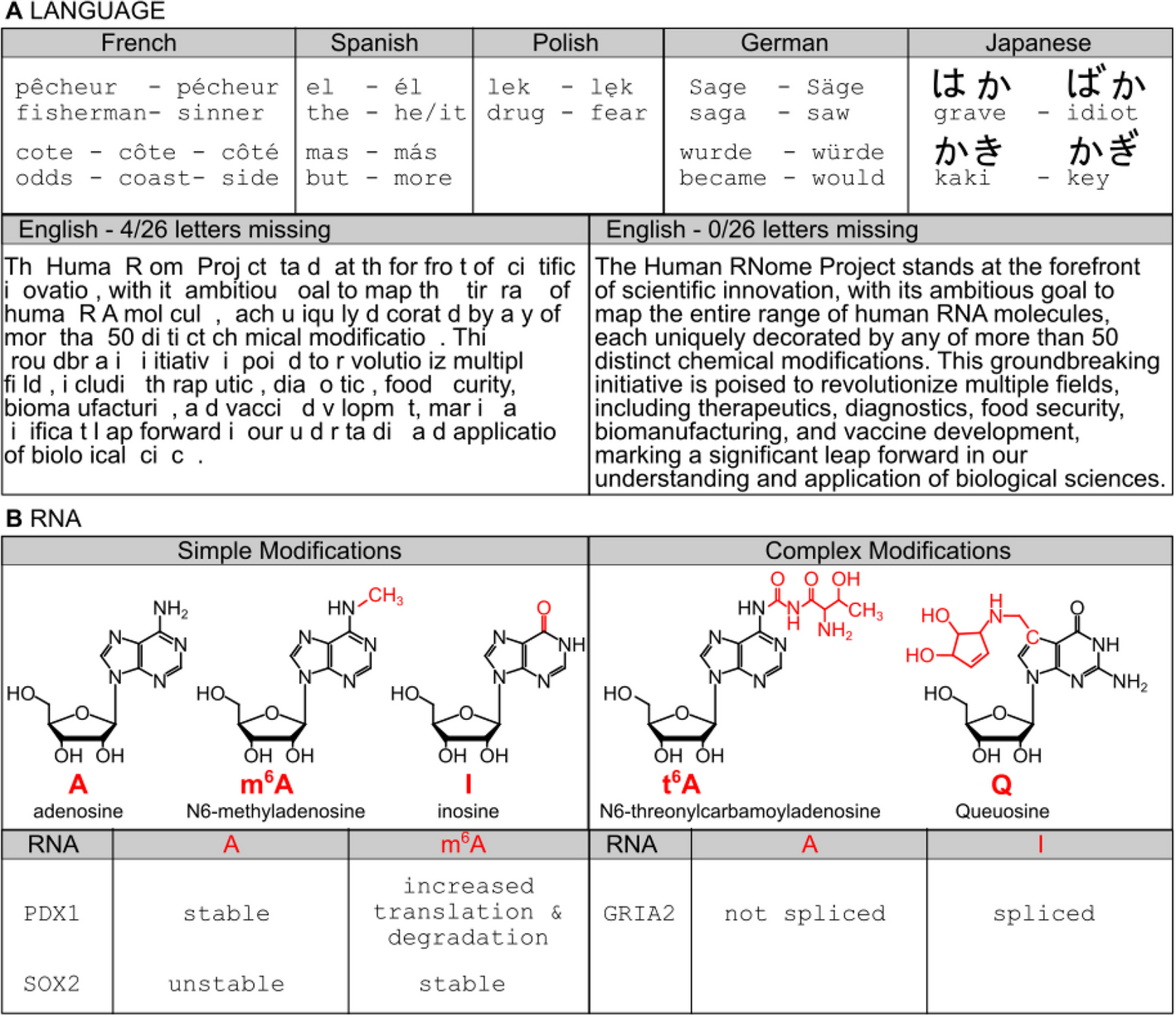
The Human RNome Project aims to ensure consistent and reproducible outcomes in RNA sequencing and modification studies by utilizing standardized cell lines maintained under uniform culture conditions. This standardized approach will facilitate meaningful comparisons across technologies and laboratories. The selected cell lines will be widely accessible, easy to maintain in culture, and highly proliferative, ensuring an adequate supply of RNA for sequencing and characterization experiments. Importantly, these cell lines will exhibit genetic stability, characterized by a well-defined genome with minimal mutations and chromosomal aberrations, to guarantee the reliability and robustness of the generated data.
To maintain genomic integrity, cell lines will be sourced from certified distributors at regular intervals and used at low passage numbers (< 8). Genetic integrity will be independently verified through DNA and cDNA sequencing, with results reported alongside direct RNA sequence data. This ensures that any genomic drift is identified and accounted for in downstream analyses.
Table 1 lists cell lines that meet these criteria. These lines have been extensively characterized by large-scale studies such as the ENCODE Project [25, 26] and the 1000 Genomes Project [17, 18]. For instance, GM12878, a cultured B-cell line from a female donor with ancestry from Northern and Western Europe, has been sequenced as part of the 1000 Genomes Project and characterized by ENCODE. IMR-90 lung fibroblasts, BJ foreskin fibroblasts, and H9 human embryonic stem cells are similarly well-characterized and available through trusted sources like Coriell, ATCC, and WiCell, which will also enforce standardized protocols for culturing and handling. Given the sensitivity of RNA to environmental factors, these standardizations are critical for ensuring data comparability. Repositories will also require users to follow consistent protocols for culturing and RNA extraction, as variations in these processes could influence RNA sequence and modification profiles.
RNA extraction and quality control: RNA will be extracted using a guanidinium thiocyanate-based method to ensure high purity and integrity. RNA quality will be assessed by absorbance ratio (260/280 and 260/230 nm) and capillary electrophoresis (e.g., Agilent TapeStation), requiring a minimum RNA Integrity Number (RIN) of 9 for RNA extracted from cell lines (as the project advances, and RNA samples are extracted from tissues, a lower RIN threshold such as 8 may be necessary). Aliquots of RNA will be archived for validation and further analyses.
Initial RNA targets for sequencing: The pilot phase of the Human RNome Project will focus on sequencing transfer RNA (tRNA), ribosomal RNA (rRNA), and mRNA, with a focus on selected protein-coding transcripts. These RNA classes are ideal initial targets due to their ubiquity, existing knowledge of their modification profiles, and robust expression across cell types.
tRNA (~ 250 expressed isodecoders) and rRNA (5S, 5.8S, 18S, 28S) are universally expressed and highly conserved, with well-studied modification types and locations [12,13,14]. Table 2 lists examples of modifications typically found in human mRNA, tRNA, and rRNAs. Both total tRNA and rRNAs can be purified from total RNA using electrophoresis or size-exclusion chromatography [28], while affinity-based methods such as chaplet chromatography [29] or reciprocal circulating chromatography [30] can be used to enrich for specific tRNA sequences. One drawback of all RNA purification methods is co-purification of non-target RNAs due to similar size or hybridization to target RNAs. Mass spectrometric analysis of modified ribonucleosides in purified RNA must always be viewed with suspicion for modifications found in multiple forms of RNA (e.g., m6A, m5C).
Selected protein-coding genes include ACTB, CDKN2A, ISG15, and SOD1. These genes were chosen based on their known association with diseases, moderate to high expression levels, relatively short transcript lengths (~ 1 kb), and known modifications. For example, SOD1 is associated with amyotrophic lateral sclerosis [31], while ACTB is widely expressed and associated with dystonia (Table 3).
Coding RNA enrichment methods: To detect low-abundance modifications, enriched RNA samples are critical. Initial poly-A RNA enrichment can be achieved using oligo-dT kits from various vendors [33]. For specific RNAs, biotinylated antisense oligonucleotides allow ~ fivefold enrichment [34], while microbead-based antisense oligos are claimed to achieve a 100,000-fold enrichment [35]. DNA nanoswitches offer another option, with ~ 75% recovery and purities exceeding 99.8% for RNA ranging from 22 to 400 nts [36].
Standardized RNA extraction using guanidinium thiocyanate.
Enrichment of test RNAs using antisense-based methods.
Mass spectrometry-based direct RNA-seq for short-read identification of modifications and nanopore sequencing for long-read sequencing and modification mapping.
Sequence transcriptomes from cell sorting-enriched samples of defined cell types.
Compare data with existing programs (e.g., GTEx).
Expand sequencing to include different cell types and tissues from individuals of all ages and ethnicities.
Sequence RNAs from specific subcellular regions (e.g., nucleus, cytoplasm, mitochondria).
Integrate single-cell transcriptomic and subcellular data.
The Human RNome Project relies on robust molecular resources and chemical standards to develop and validate sequencing and mass spectrometry (MS) technologies. These resources encompass synthetic and native RNA standards, as well as their building blocks, such as ribonucleosides, ribonucleotide triphosphates (NTPs), and oligoribonucleotides. High-quality standards are essential for ensuring accurate analysis of RNA modifications, their chemistry, and their precise locations within RNA molecules.
Chemical standards are indispensable for training and validating analytical methods before analyzing native RNA samples. They ensure reproducibility, correct identification of RNA modifications, and calibration of detection systems. Standards are summarized in Fig. 2 and include the following.
Overview of types of chemical standards needed for the Human RNome Project
Chemical standards for individual ribonucleosides are essential for characterizing RNA modifications and quantifying their abundance. Approximately 90 ribonucleoside standards are commercially available, with additional variants synthesized by academic laboratories. Comprehensive lists of vendors are provided on the RNome website [33], while PubChem offers detailed vendor information and links to chemical resources. Prices for these standards range from $20 to $1500 per milligram, with custom synthesis for rare modifications costing between $10,000 and $20,000. For qualitative analysis, 1 mg of a standard is typically sufficient. For quantitative analysis, we recommend assessing the purity of the standard by quantitative NMR prior to preparing calibration solutions for, as an example, LC–MS analysis. Despite the availability of over 90 modified ribonucleosides, many human-specific RNA modifications remain inaccessible as commercial standards. Furthermore, the chemical stability (shelf life) of ribonucleosides is not well-documented. For example, m1A undergoes Dimroth rearrangement to m6A during RNA processing and storage in aqueous solution [37, 38] highlighting the need for further research into ribonucleoside stability.
Ribonucleotide triphosphates (NTPs) are essential for in vitro transcription to synthesize RNA molecules longer than 20 nucleotides with defined modification profiles. Canonical NTPs are widely available from commercial sources, including isotopically labeled variants, while modified NTPs for specific ribonucleosides can also be obtained. However, these modified NTPs require rigorous verification of their chemical identity and purity, typically through techniques such as thin-layer chromatography (TLC) or LC–MS [39, 40]. In vitro transcription allows random, but not site-specific incorporation of modified NTPs [41].
Site-specifically labeled RNA oligonucleotides, ranging from 5 to > 60 nucleotides, are essential for training nanopore base callers and validating LC–MS methods. Solid-phase chemical synthesis is commonly used to produce labeled oligonucleotides and vendors typically provide mass spectra to confirm the overall product length, failure sequences, and impurities. However, comprehensive validation, such as mass spectrometric sequence verification and ribonucleoside LC–MS for modification identification, is rarely included but essential for robust validation. To ensure accuracy, researchers must advocate for detailed validation data, including MS sequence validation and ribonucleoside-specific quantification, alongside the standard mass spectra provided by vendors. Despite these advancements, the site-specific incorporation of modifications into long RNA sequences (> 60 nucleotides) remains a significant challenge [42]. Current approaches, which involve combining chemical synthesis, transcription, and ligation, are labor-intensive, low yielding, and not easily scalable. New approaches to long RNA synthesis are needed to facilitate the generation of site-specifically modified RNAs that mimic biological molecules.
Stability data for modified ribonucleosides is scarce, highlighting the need for systematic studies on shelf life.
Consistent preparation, validation, and distribution protocols are essential to ensure data comparability over time. Quality control samples must be maintained and shipped with detailed documentation.
Researchers should demand comprehensive validation data (e.g., MS/MS, sequence confirmation) from vendors to avoid errors in downstream analyses.
Sequencing and MS methods must be regularly validated using both synthetic and native standards
High-quality library of modifications with comprehensive validation data and shelf-lives. Many RNA modifications lack synthetic standards, necessitating collaboration with organic chemists for their production.
Sequencing technologies will be pivotal to the Human RNome Project, much like they were for the Human Genome Project. To evaluate the potential impact on the project, it is essential to analyze the current state and project developments over the next 5 to 10 years. The consortium has hence identified and discussed lead questions that concern the type of currently available sequencing technologies, the necessary developments in the near future, and critical quality controls.
Current methods to map modifications can be classified into direct, such as mass spectrometry or direct RNA sequencing, and indirect, which usually relies on sequencing by synthesis, wherein RNA is converted to cDNA via reverse transcriptase [16]. Both indirect and direct RNA sequencing methods require additional steps to assign modifications. This section is meant as a brief summary and not a comprehensive review of all current variations and developments (for a comprehensive review please refer to the Report by the National Academies of Sciences, Engineering and Medicine [23]).
Sequencing of cDNA, acquired through reverse transcription of RNA and analyzed through Illumina (and sometimes PacBio or Nanopore), is currently the most widely used form for indirect RNA sequencing. However, it cannot directly detect non-canonical ribonucleotides. Workarounds to map modifications rely on changing the RNA or cDNA product on a molecular level (“molecular input”) and include reverse transcriptase-based error profiling, chemical or enzymatic derivatization, and modification-specific immunoprecipitation [43, 44]. Molecular input methods utilize computational algorithms that infer RNA modifications from misincorporations, gaps, or reverse transcription arrests or reverse transcription incorporation of structurally similar bases. While powerful, no single molecular input method can comprehensively identify all modifications, necessitating the use of multiple techniques on the same RNA sample.
Oxford Nanopore Technologies is the only widely available platform currently providing protocols for direct, long-read sequencing of RNA molecules, eliminating the need for cDNA conversion and preserving endogenous or synthetic exogenous RNA modifications (Fig. 3) [45,46,47,48]. Advances in machine learning models have led to more accurate basecalling and lower error rates for sequencing full-length native RNA transcripts [47]. By analyzing unique changes in electrical currents from the direct RNA sequencing process, RNA modifications can be tentatively identified [48]. Identification of modified nucleotide residues can be achieved by comparison against unmodified control samples [49], with base-calling algorithms or supervised models that have been trained on data with known modifications [47, 48, 50]. The training of such models can be achieved using data from cDNA-based approaches, data from modification-free control samples [49], in vitro transcription-generated data, or data from synthetic RNAs. However, the generation and availability of such data and the lack of RNA modification standards still limit the number of modifications that can currently be confidently detected and identified. Furthermore, not all reads from direct RNA-seq correspond to full-length RNAs and challenges remain to detect RNA modifications that occur at the 5′ ends of RNA molecules. To overcome these barriers, researchers are actively developing “molecular input” approaches—such as introducing chemical or enzymatic treatments which change the RNA molecule—to amplify or clarify the signals associated with RNA modifications [51].
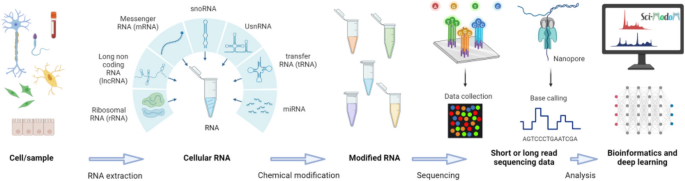
Overview of the sequencing workflow that will allow end-to-end sequencing of RNA including its modifications
Mass spectrometry (MS)-based RNA sequencing is an essential complement to these efforts as a means to chemically identify and accurately quantify specific modifications [52,53,54,55]. Unlike the chemically nonspecific interpretation of electrical signals in nanopore sequencing, MS-based sequencing involves high mass accuracy (i.e., exact molecular weight) determinations of modification fragments that allow structural identification of the modification, its location in the RNA sequence at single-nucleotide resolution, and its abundance in the population of RNA sequences. While MS sequencing requires larger quantities of RNA than NGS or nanopore sequencing, advances in sensitivity have moved the application from more abundant non-coding RNAs to mRNAs [55,56,57,58]. The major limitation of MS-based RNA-seq is the short fragment size needed for accurate MS analysis, typically 10–60 nt in length depending upon the mass resolution of the instrument [55]. This precludes mapping modifications in long native RNA molecules, as can be achieved with nanopore. MS-based RNA sequencing and nanopore sequencing are thus complementary tools for RNome analysis.
The accuracy of epitranscriptomic analysis is determined by the combined impact of errors introduced during experimental procedures and data processing. To ensure high-quality data, experimental design must include an adequate number of replicates, sufficient sequencing depth, and the incorporation of both positive and negative controls. Method-specific data analysis should employ robust statistical frameworks to evaluate the significance of signals at specific sites, accounting for sample size, signal strength, and their relevance within the broader context of all samples, including replicates and controls. Given the diversity of current modification mapping methods, it is challenging to recommend a universal set of parameters for experimental design and data analysis. Therefore, we outline guidelines based on general principles in the following sections.
Base-calling accuracy in Illumina sequencing data typically has an error rate of 0.1–0.5% per nucleotide residue, while nanopore sequencing has only recently reduced its error rates to the single-digit range. While these error rates are not typically a major concern for conventional RNA sequencing, they become critical when using molecular input methods that depend on errors for mapping modifications, as these methods can introduce artifact-based errors, such as false positives and false negatives. To ensure data validity and reliability, it is essential to include a sufficient number of both biological and technical replicates, as well as adequate sequencing depth to optimize the signal-to-noise ratio. The significance of a detected signal is further strengthened by comparisons with positive and negative controls, ideally including at least one of each that represents a “gold standard” or ground truth.
Quality control (QC) parameters are essential at multiple levels, including raw data (e.g., fastq files used for downstream analysis) and the analytical pipelines used for mapping modified residues. For raw data, QC criteria can often follow established standards for the respective sequencing technology, such as a Q-score > 30 for Illumina sequencing. The thresholds, however, may vary depending on whether short-read or long-read sequencing technologies are employed. Beyond this, a second layer of QC is needed to evaluate the performance of molecular input methods, which introduce their own characteristic errors. A third layer of QC pertains to computational analysis, assessing the reliability of data interpretation across different epitranscriptomics mapping protocols and pipelines. In some cases, it may be valuable to integrate these QC layers into aggregated error rates or composite metrics that encompass both molecular and computational aspects.
To advance the field, it is imperative to establish a universally accepted set of QC parameters for benchmarking methods. Equally important is the determination of standardized threshold values for these parameters, which could become mandatory for the Human RNome Project. The diversity of existing technologies, as well as those that will emerge during the project, complicates the establishment of universal QC criteria at the raw data level. However, any method must undergo rigorous validation before being deemed suitable for modification calling.
Validation should involve the creation of models evaluated with metrics such as receiver operating characteristic (ROC) curves, area under the curve (AUC), sensitivity (true positive rate), and specificity (true negative rate). Particular attention must be given to minimizing false positive and false negative rates, as these directly impact the reliability of modification detection. Another critical input parameter for these models is an accurate estimate of the expected number of residues for a given modification, as this will influence thresholds for modification calling. Such an integrative and standardized approach to QC will ensure robust and reliable results across diverse epitranscriptomic applications.
Establishing clear guidelines for reporting QC metrics in publications and data repositories is essential for fostering reproducibility and confidence in results. Comprehensive reporting of raw data quality, molecular input performance, and computational reliability will enable consistent practices across studies. Such transparency not only ensures accountability but also facilitates meta-analyses and comparisons, accelerating progress in the field.
Advancing modification-aware RNA sequencing on an international scale requires both organizational and technical developments. One of the greatest challenges will be achieving consensus within the field on a mandatory set of QC parameters and, even more challenging, establishing universally applicable threshold values. As highlighted earlier, in addition to maintaining a continuously updated overview of methodologies, the field must identify techniques that either deliver the highest throughput with minimal error rates or enable precise quantification of modification levels at specific RNA sites. With these considerations in mind, we outline the following ongoing and future objectives for the Human RNome Project.
Continue developing NGS, nanopore, and MS technologies to (a) expand the repertoire of modifications for NGS and nanopore by developing and refining chemical derivatization methods; (b) expand training datasets and algorithms for nanopore; and (c) increase the sensitivity, LC resolution, and data processing algorithms for MS-based sequencing.
Integrate orthogonal technologies (e.g., combinations of methods providing different molecular inputs or alternate sequencing technologies) to confirm RNA modifications with high confidence on native RNAs.
Develop and implement robust quality control (QC) protocols for NGS, nanopore, and MS to (a) minimize artifacts, (b) increase statistical power, (c) increase sequencing depth, and (d) assure inter-laboratory consistency.
Lay the groundwork for scaling and throughput: (a) multiplexing MS-based sequencing; (b) automation of sequencing library preparation, sample analysis, data processing, and data mining; and (c) inter-laboratory validation.
Create user groups to develop, implement, and cross-validate RNA-seq methods. Begin developing or adapting websites and databases for public access to protocols and RNA-seq datasets. Engage international funding bodies to support research and development.
Develop automated systems for RNA extraction, size- or sequence-based RNA purification, library preparation, sequencing, data processing, and data analysis.
Develop and refine computational methods: (a) algorithms to interpret raw sequencing data, distinguish true modification signals from noise, and quantify modifications; (b) standardize modification-calling pipelines with open datasets; and (c) develop rigorous benchmarks to ensure reproducibility.
Expand scale of sequencing efforts: Prioritize high-throughput, automated solutions to handle increasing data demands; integrate methods with lower error rates and reliable quantification into streamlined workflows.
Expand RNA-seq analyses using cell and RNA targets identified in section I. Apply improved workflows to diverse cell types and RNA populations to create comprehensive modification maps.
Develop new sequencing technologies: (a) design new nanopore pore systems, (b) develop RNA-customized MS ionization, fragmentation, and detection hardware; (c) innovate platforms capable of directly sequencing full-length RNA molecules with single-base resolution and error rates < 0.1%.
Continue developing AI and automation technologies: Design AI-driven base-calling algorithms for real-time error correction and precise modification detection.
Expand RNA-seq databases and integrate across databases.
Expand application of RNA-seq technologies: (a) cells beyond those initially identified as standards for the Human RNome Project (Section I); (b) tissues from animal models; and (c) human clinical samples.
A tremendous amount of the epitranscriptome sequencing data generated in the last few years has, on the whole, remained unused, because of limited data accessibility, poor findability and reusability. Addressing these gaps could significantly enhance the utility and impact of these data. In this section, we propose FAIR guidelines [59] for data format specifications, model training standards, and protocols for recording and sharing information related to RNA sequences and modifications, applicable to both indirect and nanopore direct RNA sequencing (Fig. 4).
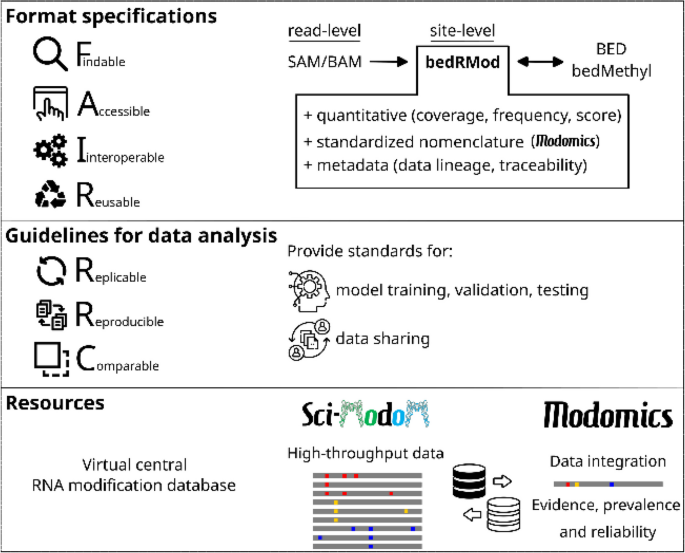
Data handling to ensure long-term and reproducible usage of the data acquired for the Human RNome Project
The identity, position, and frequency of RNA modifications are derived from large volumes of raw data, typically mapped reads. These analyses depend on method-specific technological expertise, which can vary significantly across approaches. Raw data alone are often not practical when only site-specific modification information is required. Moreover, this information has historically been disseminated through a range of incompatible formats, governed by varying standards, and often accompanied by limited access or incomplete metadata. These challenges have hindered reproducibility and the ability to compare results across studies.
While data formats for raw sequencing data are well-established, no such standardization exists for modification information derived from these data. At a minimum, site-specific RNA modification data should be reported in a straightforward format and include:
A standardized naming convention for the modification type.
Stoichiometric information, such as the percentage or frequency of the modification.
Depth of coverage for the modification site.
Quantitative confidence scores indicating the reliability of the modification call.
At the dataset level, metadata should be sufficiently detailed to ensure traceability, reproducibility, and reusability. This requires reliance on standardized nomenclature while maintaining flexibility to include free-text information where necessary. Some of this information has recently been incorporated into the latest SAM/BAM format specifications [60], where nucleotide residue modifications and their quality scores are recorded per-read.
At the per-site level, the recently proposed bedRMod format addresses many of these requirements [32]. This format is analogous to the ENCODE bedMethyl standard [61] and nanopore’s extended bedMethyl format [62] and compatible with the widely used BED (Browser Extensible Data) format. It was developed during the Human Genome Project [60] and approved by the GA4GH Standards Steering Committee and it integrates seamlessly with many command-line tools and genome browsers.
However, a significant barrier to the widespread adoption of the bedRMod format is its dependency on information of nucleotide residue modification from SAM/BAM files, which in turn relies on mapping algorithms. Tools to compile this data into bedRMod format at the site level remain underdeveloped, and current workflows often rely on custom algorithms to extract site-specific information into similar tabulated formats. Addressing this gap with robust, standardized tools will be essential to advancing the use and utility of bedRMod for RNA modification studies.
The RNA modification nomenclature adopted by MODOMICS [12, 13] aligns well with the requirements of the bedRMod format by providing standardized names for RNA modifications. MODOMICS uses a variety of representations, including multi-character alphanumeric codes for single and multiple sequence formats like FASTA, a one-letter Unicode-based code for sequence alignments, and a human-readable alphanumeric code for broader accessibility. Recent updates to MODOMICS have expanded its nomenclature to include synthetic residues, accommodating the growing diversity of RNA modifications in both research and practical applications [12]. This system is instrumental in ensuring compatibility across tools and datasets and holds potential as a foundation for a future standardized nomenclature under IUPAC guidelines.
Development of RNA modification calling algorithms requires independent datasets for model training, (cross-) validation, and additional datasets for testing and benchmarking established methods. The previous sections have outlined potential biological and synthetic sources, as well as sequencing approaches, for generating these datasets. To ensure relevance, datasets may need to align with the specific focus of interest, whether tRNAs, mRNAs, or rRNAs, as these classes differ significantly in their epitranscriptomic properties and sequence characteristics.
Additionally, datasets should accurately represent the real-world distribution of modified versus unmodified nucleotides the model is expected to encounter. They must approximate the size and complexity of existing transcriptomes and include high-quality annotations for modification classes. For example, modification-free transcripts from in vitro transcription of cDNA derived from six immortalized human cell lines have been used as a robust ground-truth dataset for unmodified mRNA transcriptomes [49]. Similarly, Chan et al. [63] employed random ligation of RNA oligos with known modification statuses to construct longer transcripts with sufficient complexity in both nucleotide composition and modification density, representing another valuable resource for RNA modification research.
The Human RNome Project seeks to establish guidelines for formatting and sharing RNA sequences and modifications, while also consolidating and integrating the growing volume of high-throughput epitranscriptome data. This effort aims to enhance data accessibility, facilitate the automated discovery of datasets, and optimize data reuse. Sci-ModoM [32] introduces a novel, quantitative framework supported by the bedRMod format, advancing the adoption of FAIR data principles and fostering the use of common standards. These features position Sci-ModoM as a potential cornerstone database for RNA modifications.
Developed in synergy with MODOMICS [12, 13], Sci-ModoM complements this meta-database by offering high-throughput, high-resolution data in a standardized format. Sci-ModoM serves as a centralized platform where modifications from diverse studies can be accessed and compared, while MODOMICS provides a curated repository of RNA sequences enriched with all known modifications, along with detailed metadata on their reliability and prevalence. Together, these resources enable the visualization of modifications within RNA sequences and broaden the utility of epitranscriptome data for research, therapeutic development, and experimental applications. The integration of Sci-ModoM and MODOMICS represents a significant milestone in achieving comprehensive annotation and effective utilization of RNA modifications.
To establish bedRMod as the format for sharing RNA modification data, and to develop the necessary infrastructure and tools to improve interoperability, and to facilitate its use by the community.
To establish guidelines and minimal requirements for training, validation, testing, and sharing RNA modification data and software.
To make realistic training and validation data available through Sci-ModoM to support the development of new detection methods and algorithms (cf. Future goals of section III).
To continuously and dynamically annotate novel modifications from the large amount of data available in Sci-ModoM using MODOMICS evidence levels, reliability scores, and prevalence metrics.
To enhance the interpretability of transcriptome-wide data accumulated in Sci-ModoM by contextualizing it with broader biochemical, structural, and functional information available in MODOMICS, bridging experimental findings with mechanistic insights.
To provide global mirroring and public access, e.g., through collaboration with academic institutions, following the open-access model of Sci-ModoM.
To build on the synergistic development of Sci-ModoM and MODOMICS to establish a virtual central RNA modification database.
To establish a standardized data flow allowing users to transition seamlessly from experimental data in Sci-ModoM to comprehensive annotations in MODOMICS.
The Human RNome Project is a bold and transformative initiative poised to revolutionize diverse sectors, including biomedicine, agriculture, data storage, and global security. By advancing RNA science, this project will deepen our understanding of RNA biology and catalyze groundbreaking innovations, delivering profound societal benefits.
Biomedicine: RNA research has made critical contributions to healthcare, particularly in understanding the biology of RNA viruses like SARS-CoV-2 and other infectious agents. These insights have accelerated the development of RNA-based therapeutics, including mRNA technologies now being adapted for applications such as influenza [64, 65] and malaria prevention [66]. Beyond infectious diseases, RNA-based therapies are transforming treatment paradigms for various conditions. Nusinersen, an antisense oligonucleotide therapy, has significantly improved outcomes for children with spinal muscular atrophy, enabling them to achieve developmental milestones [67, 68]. Inclisiran, an RNA interference-based drug, provides an effective biannual treatment for lowering LDL cholesterol, improving compliance compared to daily regimens [69, 70]. RNA-based therapies continue to advance in oncology, rare diseases, and other fields. The Human RNome Project will support this progress by improving targeting with highly accurate RNA sequences, reducing costs through the production of affordable, high-quality ribonucleotides, including modified forms, bolstering supply chains, and expanding access to RNA therapeutics.
Agriculture: Global food insecurity is a pressing issue affecting millions worldwide. In the USA alone, over 10 million children face hunger, and globally, malnutrition affected 27 million children in 2022. RNA-based technologies offer innovative solutions to address these challenges. Research shows that RNA modifications can enhance crop yields in staples like rice and potatoes [71], improving resilience and productivity. Additionally, RNA interference (RNAi) delivered via high-pressure sprays provides an effective, non-genetically engineered method to combat plant diseases [72, 73]. RNA sequencing technologies will equip plant scientists with powerful tools to improve crop productivity and combat global hunger.
Data storage: RNA presents a transformative approach to ultra-dense, efficient, and scalable data storage, capitalizing on its structural complexity and extensive chemical diversity. Unlike the binary 0,1 system traditionally used for data encoding, RNA’s repertoire of approximately 180 known ribonucleotide modifications vastly expands the encoding alphabet. While the binary system encodes 1 bit of information per symbol, RNA modifications encode approximately 7.49 bits per symbol, enabling a 649% improvement in compression efficiency.
This groundbreaking technology not only addresses the rapidly growing global demand for storage capacity, projected to surpass available resources in the coming decades, but also offers a sustainable and cost-effective alternative. By leveraging RNA’s ability to store dense information in a biochemically compact format, this innovation has the potential to revolutionize data storage while reducing the environmental and financial costs associated with conventional methods.
The Human RNome Project will be at the forefront of this innovation, establishing the necessary infrastructure to produce standard and modified ribonucleotides as foundational components for RNA-based storage. In parallel, the project will drive advancements in sequencing and synthesis technologies to ensure data integrity, reliability, and affordability. These efforts will transition RNA-based storage from theoretical concept to practical reality, opening a new frontier in data technology. By combining unparalleled storage density with innovative compression strategies, RNA-based systems promise to redefine how we store and access the world’s growing digital archives.
Pandemic and biowarfare preparedness: RNA viruses are responsible for nearly half of infectious diseases, including influenza, Ebola, hepatitis A, and COVID-19. Their high mutation rates, up to five times that of DNA viruses, make early detection and control challenging [74, 75]. The Human RNome Project will revolutionize RNA virus sequencing, enabling rapid and accurate identification of emerging pathogens. This capability will enhance global pandemic preparedness, providing the tools needed to respond swiftly to new threats. Moreover, advances in RNA sequencing will be critical for detecting engineered viruses, strengthening defenses against biowarfare, and ensuring global health and security. By enabling rapid, precise detection, the RNome Project will play a vital role in safeguarding against both natural and human-made threats.
In conclusion, the Human RNome Project will drive transformative progress across a range of fields, from health and agriculture to technology and security. By unlocking the full potential of RNA science, this initiative will deepen our understanding of fundamental biological processes and empower innovative solutions to some of humanity’s most pressing challenges. With its far-reaching applications and societal impact, the Human RNome Project promises to be a cornerstone of twenty-first-century innovation, paving the way for a healthier, more resilient future.