Tissue sample collection
Ethics approval, informed consent and experimental protocol was approved by the ethics committee of Shanghai Changzheng Hospital (2021SL044), and We confirm that all methods were carried out in accordance with the Declaration of Helsinki. Informed consent was provided by the patients and their relatives before harvesting intervertebral disc tissue during surgery. We also confirm that all methods relative to animal experiments were carried out in accordance with relevant guidelines and regulations. The experimental protocol was approved by the Institutional Animal Care and Use Committee of Second Military Medical University. Samples of normal intervertebral disc tissue were obtained from patients with lumbar trauma in the absence of radiological indications of degeneration, who underwent spinal fusion (Pfirrmann grade I, n = 6, age range 30 to 55 years, mean age 44 years). MRI T-2 weighted images were acquired, and the extent of intervertebral disc degeneration (IDD) was assessed utilizing the modified Pfirrmann grading system. All specimens were prepared for the isolation of nucleus pulposus cells.
Human primary nucleus pulposus cell culture
We performed NP cell isolation using a protocol rigorously validated by single-cell RNA sequencing (scRNA-seq)25. The collected NP tissue specimens were washed twice with sterile PBS solution. The NP region was isolated under microscopy, then minced and digested with 2 U/mL protease in DMEM medium (Gibco, NY, USA) for 20 min at 37 °C. Treating with 0.25 mg/ml type II collagenase (Gibco, Cat. No. 17101-015) for 4 h at 37 °C to obtained NP cells from the NP tissues. The remaining cell suspension was transferred into a 40 μm cell strainer (BD Biosciences, Franklin Lakes, NJ, USA) and subjected to centrifugation at 800 g for a duration of 5 min. The NP cells were subsequently resuspended in DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/mL penicillin, 100 µg/mL streptomycin, and 1% L-glutamine. The cell viability exceeded 90% as determined by the cell counting kit-8 (Dojindo, Tokyo, Japan). The cells were incubated at 37 °C in an atmosphere enriched with 5% CO2, with the culture medium being refreshed every 3 days. Cells at the second passage were utilized for additional experimental methodologies.
An in vitro model of NP cell degeneration was developed by administering recombinant human TNF-α (Peprotech, USA) (50 ng/ml) or recombinant human IL-1β (Peprotech, USA) (25 ng/ml) to the NP cells as previously reported26. For the overexpression of p53, a plasmid designed for p53 overexpression supplied by OBiO Technology (Shanghai) Corp., Ltd., or SCH529074 (MedChemExpress, USA), a p53 agonist, was utilized. Muscone (3-methylcyclopentadecanone) was purchased commercially with the purity over 98.0% (HY-N0633, MedChemExpress, China).
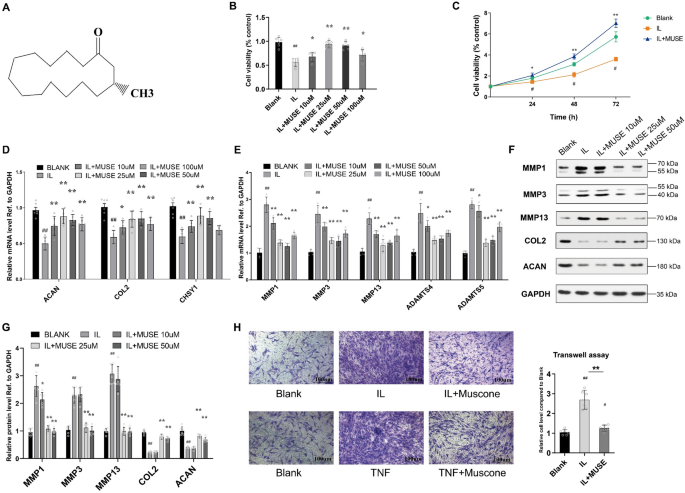
CCK-8 cell viability assay
A CCK-8 assay kit (YEASEN, Shanghai) was used to measure cell viability. Briefly, approximately 5 × 103 NP cells were seeded into each well of a 96-well plate. After treatment with the indicated group, 10 µl of CCK8 agent was added to each well according to the manufacturer’s instructions. After 1–4 h of incubation at 37 °C, the absorbances of different groups NP cells were measured with a multifunctional detection instrument (SPECTRAMAX 13X, Molecular Devices, USA).
TUNEL staining
Cell apoptosis was measured by transferase dUTP nick-end labeling (TUNEL) staining. Briefly, after different treatments, NP cells were fixed with 4% paraformaldehyde for 1 h and then cultured with 0.5% Triton X-100 in PBS for 10 min. After washing with PBS, the cells were incubated with reagents from a TUNEL Apoptosis Detection Kit (YEASEN, Shanghai) according to the kit’s instructions for staining. The nuclear counterstaining procedure was performed by incubating samples with 0.1 mg/mL 4’,6-diamidino-2-phenylindole (DAPI; Beyotime Biotechnology, Cat. No. C1002) in phosphate-buffered saline (PBS) for 5 min at room temperature. Apoptosis was observed under a fluorescence microscope (CKX41, Olympus, Japan), and photographs were obtained. At least five different fields in one sample was photographed to compare the effect.
Isolation of RNA and quantitative real-time polymerase chain reaction
RNA was isolated from human NP cells utilizing RNAiSO reagent (TAKARA, Japan) in strict adherence to the manufacturer’s protocol. Quantification of total RNA concentrations was conducted at a wavelength of 260 nm using a spectrophotometer (DU-800; Beckman Coulter, Brea, CA). Reverse transcription was performed by the PrimeScript™ RT reagent Kit with gDNA Eraser (TAKARA, Japan) in a 20-µl mixture according to the manufacturer’s instructions. TB Green® Premix Ex Taq™ (TAKARA, Japan) was used to conduct real-time PCR on a Step One Plus real-time PCR system (Applied Biosystems, Foster City, CA). GAPDH was used to normalize the mRNA expression of other genes. The comparative Ct method was used to calculate the relative expression of each transcript. Each experiment was repeated at least three times independently. The primers used in this study are listed in Supplementary Table S1.
Western blotting
Ice-cold lysis buffer (Cell Signaling Technology, Danvers, MA, USA) was used to harvest total protein from human NP cells. For measurement of phosphorylated protein levels, total protein was extracted at 12–24 h after treatment. A bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL, USA) was used to determine the concentration of protein. Protein degeneration was prevented by using PMSF (Beyotime, China) and protease inhibitor cocktail (Meilune, China). The proteins were subjected to transfer onto PVDF membranes (Bio-Rad, CA) via electroblotting. Subsequent to this, the membranes were blocked utilizing 5% non-fat milk in TBST (50 mM Tris (pH 7.6), 150 mM NaCl, 0.1% Tween-20) and subsequently incubated in TBST containing specific antibodies overnight at 4 °C. The protein bands were visualized using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, USA) and Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific, USA) to assess the expression of target proteins. The following antibodies were used: anti-MMP1 (ab52631, 1:2500 dilution), anti-MMP3 (ab53015, 1:1000 dilution), anti-MMP13 (ab39012, 1:1000 dilution), anti-ADAMTS4 (ab185722, 1:500 dilution), anti-ADAMTS5 (ab 41037, 1:250 dilution), anti-ACAN (ab3778, 1:500 dilution), anti-CHSY1 (ab153813, 1:500 dilution), anti-p53 (ab 32389, 1:10000 dilution), anti-p-p53 (ab33889, 1:1000 dilution), anti-PUMA (ab54288, 1:1000 dilution), anti-Bax (ab32503, 1:1000 dilution), anti-p21 (ab109520, 1:1000 dilution), anti-p16 (ab 108349, 1:2000 dilution), anti-CCNG2 (ab 203314, 1:500 dilution) (all from Abcam, USA) and anti-GAPDH (1:5000 dilution) (Proteintech, USA). Uncropped images were provided in supplementary Fig. 1 (Figure S1).
High-throughput RNA sequencing and bioinformatics analysis
Total RNA was isolated for the purpose of transcriptome sequencing utilizing the TRIzol reagent (Invitrogen, Carlsbad, USA), adhering strictly to the manufacturer’s prescribed protocol. Subsequently, the procedures encompassing library construction, RNA quality assurance, purification, quantification, and validation were executed by Shanghai NovelBio Bio-Pharm Technology Co., Ltd. For the analysis of the transcriptomic data, approximately 100 bp reads were aligned to the human genome (hg19) employing TopHat2/Bowtie2. The data were subsequently mapped to gene structures derived from RefSeq utilizing the summarize overlaps function in Intersect Strict mode (Genomic Ranges, Bioconductor). From the raw count data, reads per kilobase per million reads mapped (RPKM) values were computed for the identical gene set using Cufflinks. Differential expression analysis was conducted with the aid of edgeR, employing TMM (trimmed mean of M-values) library normalization and maintaining a false discovery rate (FDR) of 0.05. The datasets generated during the current study are available in the Gene Expression Omnibus (GEO) repository (GSE113633, Supplementary Dataset S1-3).
Gene Ontology (GO) and KEGG pathway analyses were performed as previously described19. In summary, the p value for each Gene Ontology (GO) term was calculated employing right-sided hypergeometric tests, and the Benjamini–Hochberg method was applied for the correction of multiple tests. An adjusted p value lower than 0.05 was considered indicative of a statistically significant deviation from the anticipated distribution, suggesting enrichment of the specific GO term or pathway among the target genes. We performed GO and KEGG pathway analyses of all the differentially expressed mRNAs and GO analysis of the mRNAs that were included in the miRNA regulatory network (Supplementary Dataset S2, S3).
Transwell invasion assay
An invasion assay was performed with a Matrigel-coated Transwell plate to assess MMP secretion from NP cells and the migration ability of NP cells after different treatments. NP cells were plated into the upper chamber, which was coated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). According to our previous study27the inflammatory cytokines IL-1β and TNF-α were administered 8 h post-plating at concentrations of 25 ng/ml and 50 ng/ml, respectively. After an incubation period of 48 h, cells were carefully removed from the Matrigel-coated surface of the upper chamber using swabs. The invaded cells on the lower surface of the chamber were fixed with 4% paraformaldehyde for a duration of 30 min followed by staining with 0.1% crystal violet for 10 min. Distilled deionized water was employed to wash the cells until clarity of the water was achieved. The samples were then allowed to air dry before being subjected to microscopic observation. Finally, the number of invaded cells were observed and counted using a microscope by selecting five different fields from one sample to generated the average cells.
Flow cytometry
Apoptotic cell measurement was conducted through flow cytometry employing an Annexin V-FITC/PI Double Staining Kit for Apoptotic Cells (BD Bioscience, USA). Cells were harvested after a 48-hour treatment period, enzymatically digested using trypsinase without EDTA, and rinsed with PBS before being processed according to the manufacturer’s protocol. Concisely, cells were resuspended in 100 µl of binding buffer, placed into a centrifuge tube, and incubated with 5 µl of dye under dark conditions for 15 min. Subsequently, 400 µl of binding buffer was added, and the samples were subjected to analysis by flow cytometry (CyAn ADP, Beckman, USA).
Cell cycle analysis
Cell cycle distribution was determined by flow cytometry using Cell Cycle Staining Kit (MultiSciences, Hangzhou). The NP cells were collected after 48 h of treatment, harvested by trypsinase without EDTA, washed by PBS, and followed up with the manufacturer’s instruction. Next, 1 ml DNA Staining solution and 10 µl Permeabilization solution were added to each tube, then incubate in dark for 30 min. Then the tube was detected by flow cytometry (CyAn ADP, BECKMAN, USA).
Senescence-associated β-galactosidase (SA-β-gal) staining
A senescence β-galactosidase staining kit (Beyotime Biotechnology, Shanghai) was used to determine the percentage of SA-β-gal-positive cells. Briefly, NP cells were cultured in 6-well plates and treated for 48 h. After fixation for 15 min, the cells were washed with PBS and then incubated with staining solution overnight. Then, the staining was observed and photographed under a fluorescence microscope (CKX31, OLYMPUS, Japan). Within each well, 500 cells were analyzed to determine the percentage of SA-β-gal-positive cells.
Animal study
Male C57BL/6 mice were purchased from Shanghai Model Organisms Center, Inc. (Shanghai, China). An anterior disc puncture-induced IDD animal model was established as described in our previous study27. Initially, ketamine (100 mg/kg) and acepromazine(3 mg/kg) was administered via intraperitoneal injections to induce general anesthesia, and local anesthesia was done by using bupivacaine prior to the incision procedure. The mice were systematically allocated into four distinct groups: the sham surgery group, the needle puncture group, the needle puncture combined with vehicle (1:2 PBS and 1:2 DMSO) injection group, and the needle puncture combined with muscone injection group. The ventral aspect of the intervertebral disc (IVD) was accessed through an abdominal incision. A needle was vertically introduced into the disc from the ventral side and rotated 180 degrees along its axis for a duration of 10 s. A 31-gauge needle was precisely inserted 1.5 mm parallel to the endplate, traversing the annulus fibrosus (AF) to the nucleus pulposus (NP), thereby compromising the integrity of the nucleus. In the control NC injection group, a vector of 10 µl was meticulously injected into the punctured IVD, after leaving the needle in place for five seconds, the needle was carefully removed without excessive mechanical injury. In the muscone injection group, an injection of 10ul MUSE (25 μm), with or without the addition of SCH529074 (2 μm). Magnetic resonance imaging (MRI) was conducted at intervals of 0, 3, and 9 weeks subsequent to the initial surgery. The spine of mice was analyzed in sagittal T2-weighted images using a 3.0 T clinical magnet (Philips Intera Achieva 3.0 MR). T2-weighted sections in the sagittal plane were obtained using the following settings: a fast spin echo sequence with a time to repetition (TR) of 5400 ms and a time to echo (TE) of 920 ms; a 320 (h) x 9256 (v) matrix; a field of view of 260; and 4 excitations28,29. The section thickness was 2 mm with a 0-mm gap28. The Pfirrmann grading system facilitated the evaluation of intervertebral disc (IVD) degeneration. ImageJ and Surgimap software were employed to examine the alterations in the IVD30.
Histopathological analysis and immunohistochemical staining
IVD sections from each group were subjected to hematoxylin and eosin (HE) staining and Safranin O-fast green (SO) staining. A histopathological grading system considering the cellularity and morphology of the AF and NP as well as the border between the two structures was used to evaluate histopathological changes31. A histopathological score of 5 indicated no degeneration, 6–10 indicated moderate degeneration and 11–15 indicated severe degeneration. Briefly, the sections were blocked with 1% (w/v) goat serum albumin at 37 °C for 1 h. Then, the sections were incubated with primary antibodies (p53, p16, p21; 1:100 dilution) overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies for 1 h at 37 °C. The samples were imaged by a Zeiss microscope (Zeiss Axio Imager A2, Carl Zeiss Microscopy GmbH, Jena, Germany).
Statistical analysis
The data are presented as the mean ± standard deviation of at least three independent experiments. GraphPad Prism 6.0 software (GraphPad Software, Inc., San Diego, CA, USA) was used for statistical analysis. The normality of the data was tested using the D’Agostino-Pearson omnibus normality test. The proportion of immune-positive NP cells in human NP tissues failed to satisfy the normality test; consequently, these data were subjected to analysis using the two-tailed Mann‒Whitney U test. Conversely, the remaining datasets met the criteria for normality and were accordingly analyzed using the two-tailed Student’s t-test for the comparison between two groups, or through analysis of variance (ANOVA) followed by Tukey’s t-test for multiple group comparisons. The threshold for significance was established at 0.05.