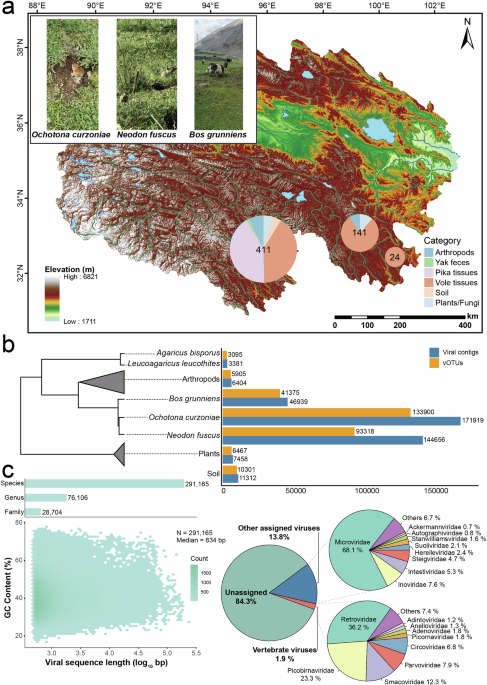
Virome of QTP small mammals and their associated environments
Virome analysis was conducted on samples from small mammals (plateau voles and plateau pikas) and their associated environments in multiple pastoral settlements across three county-level administrative regions at an average altitude of 4133 m (see Methods for details). Environment samples were collected from areas surrounding animal burrows, specifically near capture sites where burrows were visible. Notably, samples from Chengduo had the highest biodiversity, while in Jiuzhi, plateau voles were found only in barren landscapes slightly farther from pastoral settlements (Fig. 1a). A total of 392,069 viral contigs were identified across all samples following de novo assembly (Fig. 1b). After deduplication and clustering, these contigs were classified into 291,165 species-, 76,106 genus-, and 28,704 family-level viral operational taxonomic units (vOTUs). The viral species richness of these samples was assessed using rarefaction curves (Supplementary Fig. 1). As the number of sampled sequences increased, the curves gradually approached a plateau, suggesting that the sequencing depth was sufficient to capture the majority of viral diversity present in these libraries. This indicates that additional sequencing would yield only a limited number of new species. Only 15.7% of the vOTUs were successfully annotated at the family level, comprising 1.9% vertebrate-associated viruses and 13.8% other annotated viruses. Among the vertebrate viruses, the most frequently assigned families were Retroviridae, Picobirnaviridae, Smacoviridae, Parvoviridae, and Circoviridae. The other annotated viruses were dominated by Microviridae and several families within the class Caudoviricetes, such as Intestiviridae, Steigviridae, Herelleviridae, and Suoliviridae, which are mainly associated with prokaryotic hosts (Fig. 1c).
a An overview of the sampling sites in Qinghai Province, China, with three distinct sampling locations indicated by pie charts. The top-left image was taken by Qing Zhang. b The number of viral contigs and species-level vOTUs clustered from different sample categories, with the phylogenetic relationships of the samples indicated. c The distribution of 291,165 species-level vOTUs is plotted based on their length and GC content. The bar charts at the top show the distribution of these vOTUs further clustered at the genus and family levels. Dots are colored according to the number of vOTUs. The pie chart shows the proportion of well-annotated vOTUs across different classifications.
The assignment of most sequences to bacteriophages (phages) was expected (Fig. 2a). Overall, eukaryotic viruses constitute a minor component of microbial communities, yet their potential impacts are often evident19. Identifying these viruses, especially novel eukaryotic viruses, is essential for anticipating and mitigating potential future threats. We assembled 774 complete or nearly complete eukaryotic viral genomes from the majority of animal tissue and environmental samples included in this study, with nearly complete genomes being those that contain at least one annotatable conserved motif. These viruses shared sequence identity with 22 established viral families known to infect vertebrates, invertebrates, plants, and fungi, or with two groups of unclassified Circular Replication (Rep)-Encoding Single-Stranded (CRESS)-DNA viruses and unclassified RNA viruses, 95.5% of which shared less than 80% amino acid (aa) sequence identity with any known viruses (Supplementary Data 1).
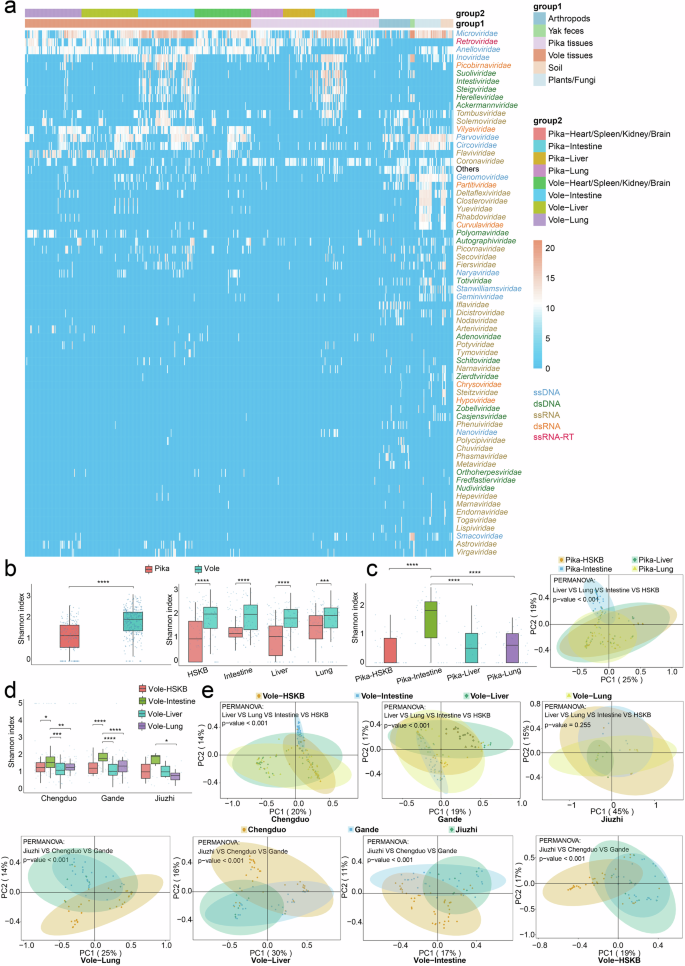
a Heatmap of viral families in the collected samples. The differently colored bands at the top of the heatmap indicate the sample sources, while the row names on the right represent the names of viral families. The data is log-transformed with a base of log10. b The left panel presents boxplots of Shannon diversity (alpha diversity) of viral communities in the overall tissues of plateau pikas and voles. The right panel presents boxplots of Shannon diversity of viral communities across four tissue types from plateau voles and plateau pikas. The Shannon index was analyzed using the Wilcoxon Rank-Sum Test. Statistical significance was defined as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; comparisons were made between the two groups. c The left panel presents boxplots of Shannon diversity of viral communities across different plateau pika tissues. The right panel shows the PCoA analysis, which revealed significant differences among viral communities from different plateau pika tissues. The two principal components accounted for 25% (PC1) and 19% (PC2) of the total variation, respectively. Each symbol represents an individual sample. d The boxplots show the Shannon index of viral communities across different vole tissues in three sampling regions. e The PCoA analysis separately illustrates the significant differences in viral communities among different plateau vole tissues across sampling sites, as well as the variations in viral communities of the four tissue types across the three sampling sites.
Virome diversity of plateau pikas and plateau voles
To gain a deeper understanding of the virome in plateau pikas and plateau voles, we constructed separate libraries for each individual’s liver, lung, intestine, and heart/spleen/kidney/brain (HSKB) samples. The heart, spleen, kidney, and brain samples were pooled into a single library, as these tissues were expected to yield very low viral loads, thereby improving detection sensitivity. Overall, the viral diversity in plateau vole tissues was higher than that in plateau pika tissues (Wilcoxon Rank-Sum Test, P < 0.0001), a consistent pattern observed across four distinct tissue types (Fig. 2b). This could be attributed to the smaller sample size of plateau pikas relative to plateau voles. Moreover, plateau pika samples were collected only from pastoral settlements in a single county-level administrative region, whereas plateau voles inhabited more biodiverse environments. Including a larger number of samples and incorporating sampling sites with greater geographic and ecological diversity may alter the currently observed virome landscape.
In plateau pikas, the viral diversity in intestinal samples was significantly higher than that in other tissues, which is expected, as the intestine is typically the most virus-rich organ in most mammals20 and humans21,22. However, there was no significant difference in viral diversity among lung, liver, and HSKB samples (Fig. 2c). Additionally, Principal Coordinate Analysis (PCoA) revealed that the viral composition of intestinal tissues was distinctly different from that of other tissues. Notably, although α diversity did not differ significantly among lung, liver, and HSKB samples, their overall viral community structures showed significant differences in β diversity analysis. This indicated that while these tissues had similar viral richness and evenness, their community compositions remained distinct.
Similarly, plateau voles from Chengduo and Gande exhibited a viral diversity pattern comparable to that of plateau pikas (Fig. 2d, e). However, in voles sampled from Jiuzhi, the distribution of viral diversity followed a different trend, with significant differences observed only between the intestine and lung tissues. Furthermore, PCoA analysis failed to distinguish the overall viral community structures among different tissues in Jiuzhi voles. Given this, increasing the sample size may help reveal a clearer diversity pattern. Additionally, we observed significant β diversity differences in the same tissue of plateau voles across different regions, which may be partially attributed to the interaction between the voles and their surrounding environments.
Highly diverse phages harbored in QTP small mammals and their associated environments
Phages are widely prevalent across diverse habitats and play a critical role in maintaining bacterial community dynamics and ecosystem stability. The majority of the annotated viruses in this study were phages (over 70%), primarily belonging to Caudoviricetes and Microviridae, while the unannotated sequences (84.3%) may also represent a substantial proportion of phages (Fig. 1c). Caudoviricetes comprise a diverse class of tailed double-stranded DNA (dsDNA) phages found in various environments and were highly represented in gut metagenomes23. To investigate the evolutionary patterns of this group in QTP small mammals and their associated environments, we generated a phylogenetic tree using a concatenated alignment of 77 protein-coding marker genes24 (Fig. 3a). After excluding genomes with insufficient data (fewer than three markers or less than 5% alignment representation), the final phylogenetic tree consisted of 4380 genus-level viral genomes, with reference genomes sourced from the most comprehensive human gut virome database, MGV25. Compared to the MGV database, this study identified 830 putative novel vOTUs at the genus level, with an average length of 36,859 bp. Notably, as more viral genomic data become available, the resolution and accuracy of detecting novel vOTUs are expected to improve. Among them, 235 were from plateau pikas, 213 from plateau voles, 187 from yak feces, 72 from arthropod samples, 62 from plant and fungal samples, and 61 from soil samples. Intestinal vOTUs were the most abundant, totaling 609 (73.4%), with 220 from plateau pikas, 202 from plateau voles, and 187 from yaks. Moreover, Caudoviricetes were not detected in the HSKB and lung samples of plateau pikas, nor in the HSKB samples of plateau voles. Additionally, the majority of the vOTUs were predicted to be assigned to host phyla Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria, while only two genomes were classified as archaeaphages. The percentage of lysogenic viruses varied across host phyla, with most exhibiting a low proportion (Supplementary Data 1). Microviridae, a family of viruses that infect bacteria, is widely recognized as one of the most diverse and globally distributed viral families26. They inhabit a variety of environments, including the guts of animals and humans27,28, insects29, freshwater30, seawater31, and sediments32. A total of 512 distinct major capsid proteins (MCPs) were identified at the genus level, with an average sequence length of 514 aa (Supplementary Data 1). Among these, 360 MCPs exhibited less than 60% aa sequence identity with their closest matches in the GenBank database. Furthermore, network clustering analysis showed that a subset of MCPs identified in this study grouped into several major clusters with known sequences, the majority of which could not be classified into specific viral genera or species. In contrast, another group of MCPs formed clusters consisting of a single or a few MCPs, suggesting novel genera (Supplementary Fig. 2). The evolutionary diversity of phages is thought to be driven by horizontal gene transfer with other microbes or host genomes33. To explore how gut viruses differ in their evolutionary relationships and functional potential among plateau voles, plateau pikas, and yaks living in the same region, we examined the relationship between nucleotide-level genetic distance and gene content similarity among viral genomes from these hosts (Fig. 3b and Supplementary Data 1). Overall, gut viruses from different hosts showed substantial nucleotide-level divergence and low gene content similarity. In contrast, the viromes of plateau pikas and plateau voles displayed greater functional similarity, suggesting a closer ecological relationship between the two species. Furthermore, the moderate gene content similarity observed between the viromes of pikas and yaks may indicate potential ecological interactions or viral exchange.
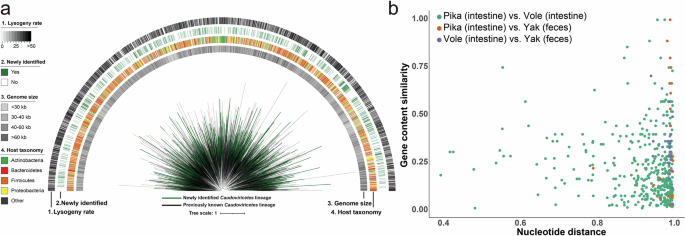
a To enhance visualization, only one representative genome is shown for each genus-level vOTU. Branch colors indicate their sources: black represents lineages from a previously published study, while green denotes those unique to this study. The outer rings display four metadata attributes for each vOTU. b The scatter plot illustrates the relationship between pairwise nucleotide distance and gene content similarity among gut viromes of plateau pikas, plateau voles, and yaks. Each dot represents a comparison between two viral genomes derived from different host species. The x-axis shows the nucleotide distance, while the y-axis indicates gene content similarity, reflecting the extent of functional overlap between viral genomes.
Phage sequence-derived genes were annotated with the KEGG search function in eggNOG-mapper v2, enabling the investigation of their potential roles (Supplementary Data 2). To focus on host-associated phages and reduce environmental interference, we restricted our analysis to those detected in plateau pikas and voles. As expected, these functional genes were predominantly detected in intestinal tissues (Fig. 4a). The results revealed that the majority of annotated genes were associated with genetic information processing, particularly its subcategory, replication and repair, with a significant portion remaining unclassified, indicating their critical roles in preserving genomic integrity and cellular functions. To enhance the visualization of relationships between these genes and host tissues, we included only genes with a weight ≥ 50 in the network diagram. Among these genes, those associated with pyrimidine metabolism (thyX and dut) and DNA replication (dnaE and polA) had the highest numbers and were detected more frequently in the intestinal tissues of plateau pikas than in those of plateau voles (Fig. 4b). Interestingly, although more plateau vole samples were included in this study compared to plateau pikas, and their intestinal tissues exhibited greater viral diversity (Fig. 2b), these genes were still present in higher numbers in plateau pikas. One possible explanation is the differences in adaptive strategies between plateau pikas and plateau voles for high-altitude adaptation.
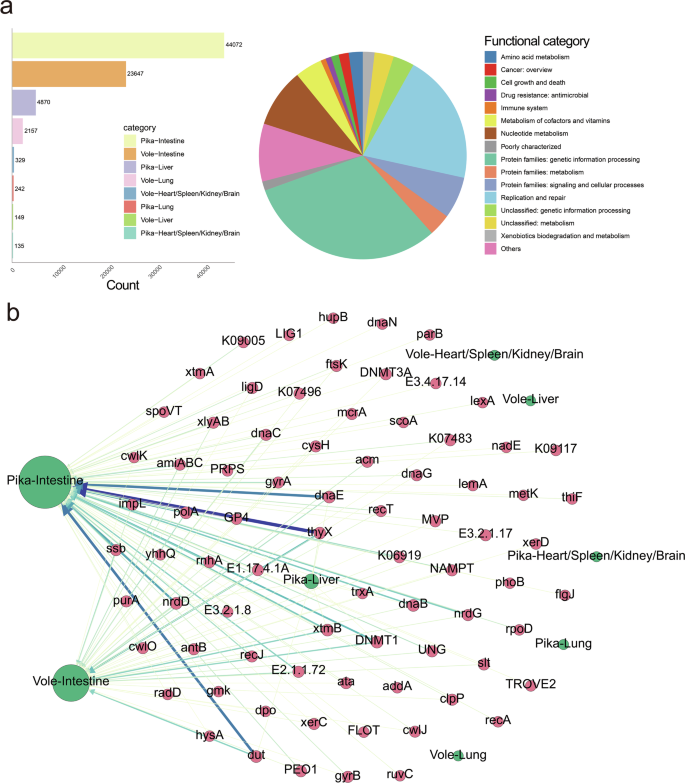
a The bar chart on the left shows the number of functional genes in different plateau pika and vole tissue categories annotated based on the eggNOG database, while the pie chart on the right illustrates the classification of these functional genes. b Network diagram of viral functional genes and their source tissues. Arrows always point from genes to their sources, with genes shown as red circles and sources as green circles. The thickness and color intensity of the arrows are positively correlated with the degree of association between genes and their sources.
Evolutionary diversification of novel eukaryotic viruses
We manually validated the genomes of 774 viral sequences and inferred phylogenetic trees based on RNA-dependent RNA polymerase (RdRp) sequences for RNA viruses, replicase or other non-structural proteins for DNA viruses, and full-length ORF1 sequences for anelloviruses (Fig. 5 and Supplementary Data 1). The 22 unedited phylogenetic trees with detailed sequence information are available in Supplementary Fig. 3.
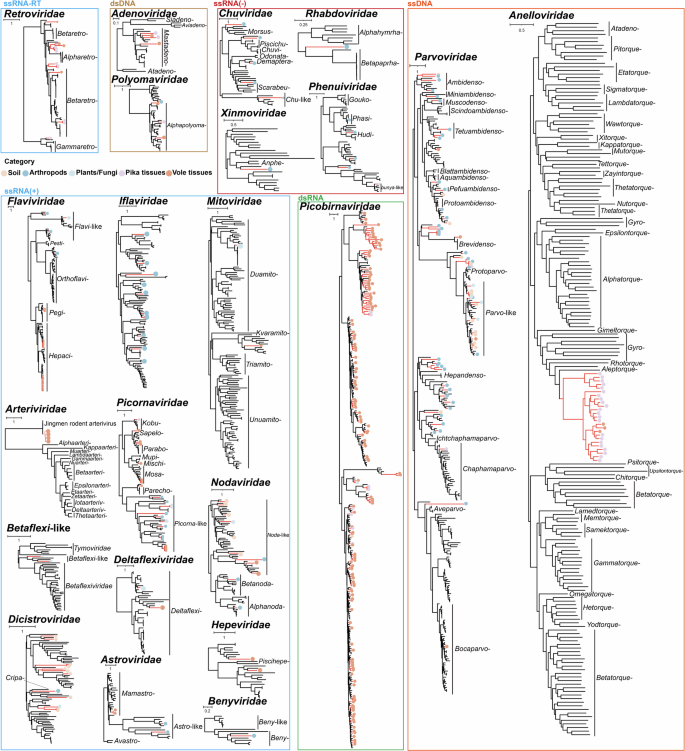
The phylogenetic trees were primarily constructed based on the amino acid sequences of RdRp for RNA viruses and replicase and other non-structural proteins for DNA viruses. Additionally, following ICTV classification guidelines, the phylogenetic tree for Anelloviridae was constructed using the nucleotide sequences of the complete ORF1.
Positive-sense ssRNA viruses and retrotranscribing RNA viruses
A total of 99 positive-sense ssRNA viruses were detected, classified into eleven viral families (Arteriviridae, Deltaflexiviridae, Flaviviridae, Astroviridae, Benyviridae, Hepeviridae, Dicistroviridae, Picornaviridae, Mitoviridae, Nodaviridae, and Iflaviridae), as well as Betaflexi-like viruses. In addition, 13 members of the Retroviridae family (ssRNA-RT viruses) were also identified. Within the Arteriviridae family, four viruses closely related to Jingmen rodent arterivirus 1 were identified in the lungs of plateau voles from Gande and Chengduo, sharing 84.3–85.3% aa sequence identity with this virus. Interestingly, viral fragments were also detected in the liver and HSKB samples of voles, suggesting potential systemic spread within the host (see the section ‘Cross-species transmission of viruses’ for details). Twenty-four viruses were classified into the Flaviviridae family, with phylogenetic analysis effectively grouping them into distinct clades based on host preference. Among them, two Pegivirus viruses shared 83.8–83.9% aa sequence identity with Phaiomys leucurus pegivirus. Additionally, 18 Hepacivirus viruses were identified as being related to rodent hepacivirus, sharing 67.4–87% aa sequence identity. Both Pegivirus and Hepacivirus viruses were detected in the liver and lungs of plateau voles. Two hepeviruses identified from the intestines of Chengduo plateau voles shared less than 55% aa sequence identity with any known viruses, suggesting the presence of novel viral species or possibly even new genera. Similarly, 24 picornavirus-related sequences were identified in the intestines of plateau pikas and voles, as well as in arthropods and fungi, indicating that these viruses occupy the ecological interface between QTP small mammals and their associated environments. Some of these sequences may represent novel species, as they formed distinct phylogenetic clades. Four astrovirus-related viruses were detected. Among them, two were identified in the intestines of plateau voles, sharing 67–68.8% aa sequence identity with their closest matches from voles in South Dakota. The other two viruses, detected in arthropod samples, exhibited low identity to known viruses and were therefore classified into the Astro-like group. Moreover, novel viruses related to Iflaviridae, Deltaflexiviridae, Betaflexiviridae, Dicistroviridae, Benyviridae, and Mitoviridae were detected, with natural hosts primarily including invertebrates, plants, fungi, and insects. However, these viruses were only sporadically identified in small mammals, suggesting that they may have a limited role in the interactions between QTP small mammals and their associated environments. In contrast, Nodaviridae appears to be more frequently involved in viral transmission between small mammals and their associated environments.
Negative-sense ssRNA viruses
A total of 12 negative-sense ssRNA viruses associated with the families Chuviridae, Rhabdoviridae, Xinmoviridae, and Phenuiviridae were identified from arthropods, plants, and fungi. Among the five viruses associated with Chuviridae, one shared 74.7% aa sequence identity with a Chuvirus previously identified in the feces of a rat from the QTP, suggesting it represents a novel species. The other four exhibited 34–45.8% aa sequence identity with their closest matches in GenBank, indicating that they likely represent four distinct novel genera34. A novel virus belonging to Rhabdoviridae was identified, with a complete genome length of 14,494 bp. It shared 36.7% aa sequence identity with its closest match, which was originally detected in Lasius neglectus. Phylogenetic analysis revealed that it formed an independent clade between the genera Alphahymrhavirus and Betapaprhavirus, suggesting that it may represent a novel genus or a higher taxonomic rank. Similarly, a virus with a full genome length of 13,447 bp was classified within the Xinmoviridae family. It shared 38% aa sequence identity with its closest match, Soldier fly-associated anphevirus, identified in Inopus flavus. Given its low sequence identity and phylogenetic analysis, it is likely to represent at least a novel species. Additionally, five viruses related to Phenuiviridae were identified. Among them, one virus shared 95.7% aa sequence identity with its closest match, while the remaining four exhibited less than 55% identity with their respective closest matches. Based on phylogenetic analysis, each of these four viruses likely represents a novel species.
dsRNA viruses
In this study, a total of 154 picobirnaviruses were identified, with 144 derived from the intestines of plateau pikas and voles, 6 from the lungs of voles, 1 from the liver of a pika, and the remaining 3 from environmental samples. These viruses exhibited aa sequence identities ranging from 25.2% to 79.8% with their most closely related sequences in GenBank, including those detected in pigs, foxes, chickens, marmots, mongooses, and cattle. Notably, the vast majority of sequences were detected in the intestines of small mammals, suggesting an association with the host’s diet. However, the relatively low number of viruses detected in environmental samples, compared with the number found in the host intestines, suggests that these viruses are primarily transmitted among mammals rather than being widely circulating in the environment.
ssDNA and dsDNA viruses
A total of 63 sequences related to Parvoviridae were identified from the intestines of plateau pikas, the intestines and HSKB of plateau voles, as well as from arthropods, plants, fungi, and soil samples. Among them, 49 sequences shared less than 70% aa sequence identity with their closest matches. Additionally, these parvoviruses span a wide taxonomic range from the subfamilies Densovirinae to Parvovirinae, suggesting their widespread distribution on the QTP. Moreover, four viruses belonging to the genus Mastadenovirus were detected in the tissues of plateau pikas and voles. Additionally, four viruses related to the family Polyomaviridae were identified from the liver and lungs of plateau voles, the lungs of plateau pikas, and arthropods. Phylogenetic analysis based on the aa sequences of the large tumor antigen revealed that these viruses clustered into distinct evolutionary clades.
Two novel monophyletic clades of Anelloviridae
A total of 25 viruses belonging to the family Anelloviridae were identified, all derived from various tissues of plateau pikas and voles. Interestingly, BLASTn searches of their ORF1 nucleotide sequences returned no significant matches. Additionally, phylogenetic analysis revealed that these viruses formed two distinct clades, comprising 5 and 20 viruses, respectively. These two clades should at least be considered to represent two distinct new genera.
Unclassified RdRp-encoding RNA and CRESS DNA viruses
We recovered 108 RdRp sequences from the dataset, which exhibit a notable degree of divergence from all currently known RdRp sequences, making it difficult to assign them to any established viral taxonomy. Sequence similarity network analysis revealed that most sequences formed distinct clusters, suggesting an evolutionary relationship with known but yet-to-be-classified viruses (Fig. 6). In contrast, some isolated RdRp sequences may represent new viral species, genera, or families. CRESS DNA viruses are widely distributed and have been detected across a wide range of eukaryotic hosts35. These viruses display remarkable genetic diversity and continue to proliferate without signs of decline36. To investigate the diversity of CRESS DNA viruses in small mammals and their associated environments on the QTP, we recovered 292 Rep protein sequences from the dataset. These sequences included 11 circoviruses, 15 genomoviruses, 4 geminiviruses, 25 smacoviruses, and 237 unclassified CRESS DNA viruses. Further sequence analysis showed that 177 Rep proteins exhibited less than 60% aa sequence identity in comparison to known viruses, suggesting that they may represent potentially novel viral species. Notably, CRESS DNA viruses exhibited distinct host specificity. Compared to other eukaryotic viruses, smacoviruses were detected at significantly higher proportions in yak feces, whereas the representation of other eukaryotic viruses in yak feces was relatively low. This finding suggests that the viromes of the yak gut and small mammals have limited ecological interactions.
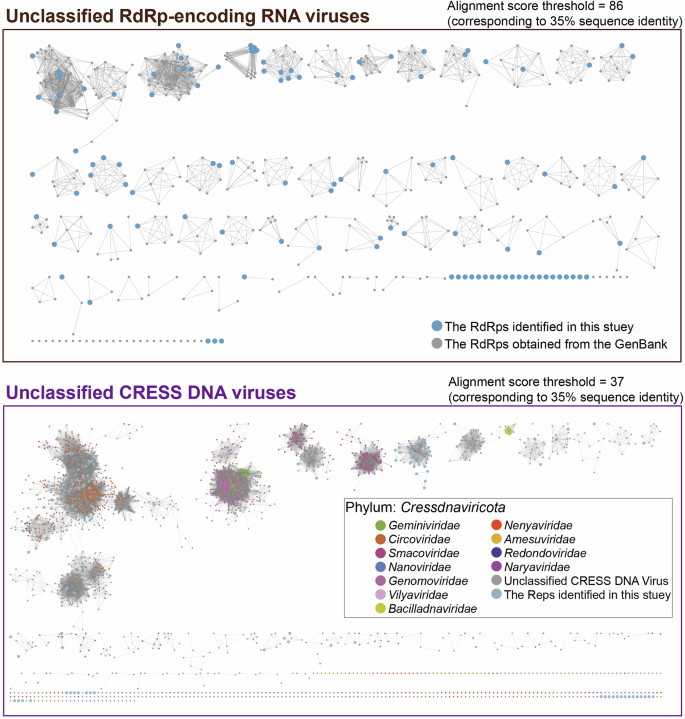
Viruses identified with RdRp / Rep sequences but unable to be classified were included in the sequence similarity network analysis along with their best matches from the current GenBank database and other representative viruses.
Tissue-associated differences in the presence and composition of eukaryotic viruses in plateau pikas and plateau voles
To better understand the distribution of eukaryotic viruses across different tissues in plateau pikas and plateau voles, we clustered sequences annotated as eukaryotic viruses using 95% ANI across 90% of the shortest assembled contig to define species-level clusters. Consequently, these viruses were classified into 14 viral families and two unclassified viral groups (Supplementary Data 3). The heatmap illustrates the distribution of different viral groups across various tissues (Fig. 7a). Notably, the unclassified Cressdnaviricota family was detected in a large proportion of samples but appeared more frequently in the intestinal tissues of voles (Supplementary Fig. 4). Additionally, as noted earlier, smacoviruses were more commonly found in yak feces. Flaviviridae was mainly present in the liver and lungs of voles, whereas Anelloviridae was found more often in pika tissues than in vole tissues. More specifically, Adenoviridae was detected only in the intestinal tissues or the HSKB of voles, with no evidence of cross-tissue transmission. It is worth noting that analyzing the heart, spleen, kidney, and brain separately when constructing libraries could provide a clearer picture of their distribution. Arteriviridae appears to be exclusively associated with voles. Additionally, Anelloviridae was widely detected across multiple tissues and individuals of both plateau pikas and plateau voles, suggesting a strong capacity for tissue-level transmission, though no clear evidence of cross-species transmission has been observed. Similarly, Circoviridae and several yet-to-be-classified viruses exhibited comparable distribution patterns (Supplementary Fig. 4). Additionally, two representative contigs from the Retroviridae family, Hvole267_72163 (2978 bp) and Hvole267_1029205 (2974 bp), were originally detected in the intestine of plateau voles and share only 32.1% nucleotide identity, yet were unexpectedly found across various tissues of plateau pikas, suggesting the possibility of cross-species transmission (Fig. 7b).
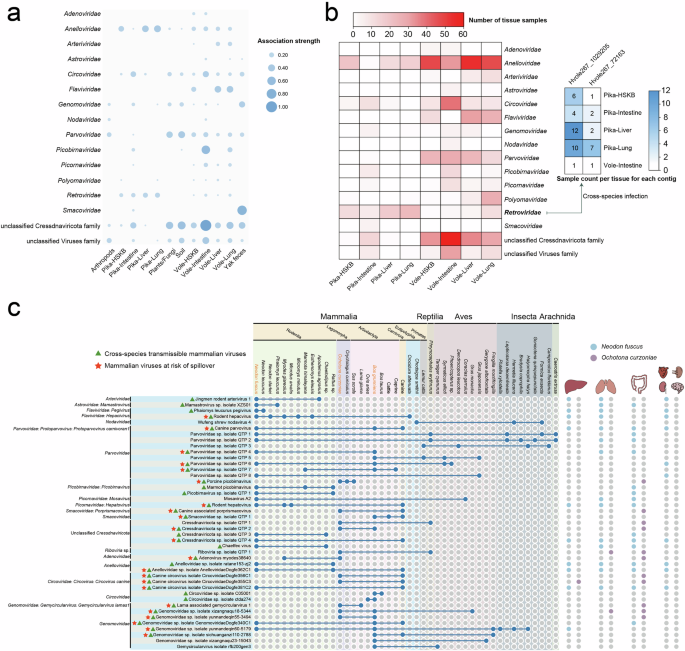
a Circle heatmap illustrating the associations between viruses and host samples (tissues). Each circle represents a virus-host association, and the circle size is positively associated with the number of viruses detected in each host sample. b The heatmap on the left shows the distribution of viral families detected in various tissues of plateau voles and pikas. Color intensity represents the number of samples in which each viral family was identified. The heatmap on the right highlights two species-level representative contigs initially detected in the intestines of plateau voles that were also identified in different tissues of plateau pikas, indicating potential cross-species transmission. c The cross-species transmission pattern of 47 known viruses was mapped based on the data generated in this study and viral records from the NCBI database. The Latin names of mammalian samples collected in this study are displayed in yellow at the top of the figure. Blue circles indicate the presence of these viruses in different hosts (see Supplementary Data 4 for details). On the right, purple and skyblue circles represent the presence of these viruses in plateau pika and vole tissues, respectively.
Cross-species transmission of viruses
To evaluate the viruses identified in this study, particularly those associated with vertebrates and their potential public health impact, we focused on viral taxa with cross-species transmission potential (detected in at least two mammalian species) and spillover risk (detected in mammalian hosts from at least two distinct orders)37. Species-level classification of known eukaryotic viruses was based on ≥80% amino acid identity in conserved proteins38,39. All known viruses included in the analysis were obtained from viral records in the NCBI database and the corresponding literature (Supplementary Data 4). We identified 32 mammalian viruses capable of cross-species transmission, 22 of which are considered at risk of spillover, including Rodent hepacivirus, Rodent hepatovirus, Canine parvovirus, Porcine picobirnavirus, and Canine-associated porprismacovirus, among others (Fig. 7c). The genomic organizations of these viruses can be found in Supplementary Fig. 5.
Notably, some viruses that had not been detected in other regions since their initial identification were also found in this study. Adenovirus myodes38640 (with spillover risk) was identified in Myodes glareolus from Ukraine; Chaetfec virus (with cross-species transmission potential) was detected in the feces of Chaetodipus sp. in Arizona, USA; and Mosavirus A2 was initially identified in the feces of Coracias garrulus collected in Hungary40. Mosavirus A2 is likely transmitted via bird droppings, raising the possibility that plateau voles may have acquired the virus through environmental exposure. In this study, however, Mosavirus A2 and Chaetfec virus were detected in the lungs and intestines of plateau voles (Supplementary Data 4). Additionally, some viruses are estimated to have spread globally, including Rodent hepacivirus, Porcine picobirnavirus, Rodent hepatovirus, Wigfec virus, Circoviridae sp. isolate ctda274, and Lama-associated gemycircularvirus 1.
We identified canine parvovirus (Protoparvovirus carnivoran1) in various tissues of plateau voles collected from three county-level administrative regions, as well as in yak feces. This virus was previously identified in canine fecal samples obtained from the QTP41. Additionally, several viruses that had been previously identified in Rodentia, including Jingmen rodent arterivirus 1, Mamastrovirus sp. isolate XZS01, Phaiomys leucurus pegivirus, Rodent hepacvirus, Rodent hepatovirus, Chaetfec virus, Anelloviridae sp. isolate ratane153-zj2, and Anelloviridae sp. isolate AnelloviridaeDogfe362C1, were further characterized in various tissues of plateau voles.