Sample Page
Solar wind ‘U-turn’ spotted by Parker Solar Probe
Written by
admin
in
7. Science
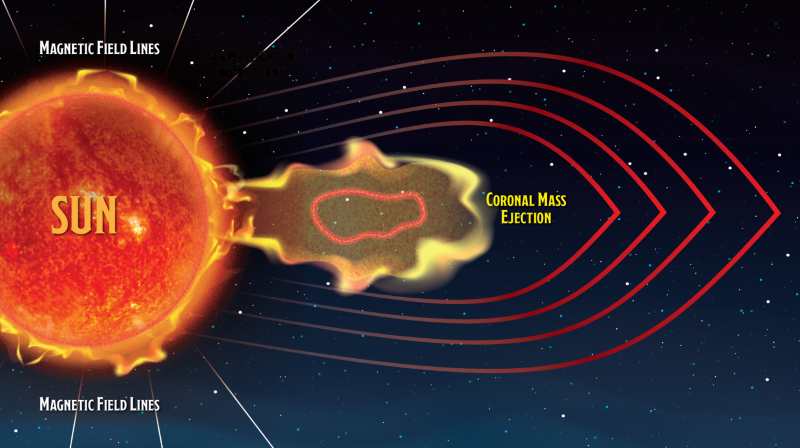
These images of our star’s solar wind, captured by NASA’s Parker Solar Probe, show a phenomenon called an inflow occurring in the sun’s upper atmosphere. This is where solar material rains back toward the sun’s surface.
Originally…
Continue Reading
←
Russia and China pledge support for Venezuela as Trump ratchets up pressure on Maduro | Venezuela
Enabling synergies between solar PV projects and the local environment – events
→
More posts
Cannon McIntosh Doubles Up in Dramatic BC39 Win
December 23, 2025
Top 5 Most-Read PHEO Articles of 2025
December 23, 2025
Jail for Wyke woman found with £8.5m stash of heroin
December 23, 2025
Full Goods Diner at Pearl to close in late December
December 23, 2025