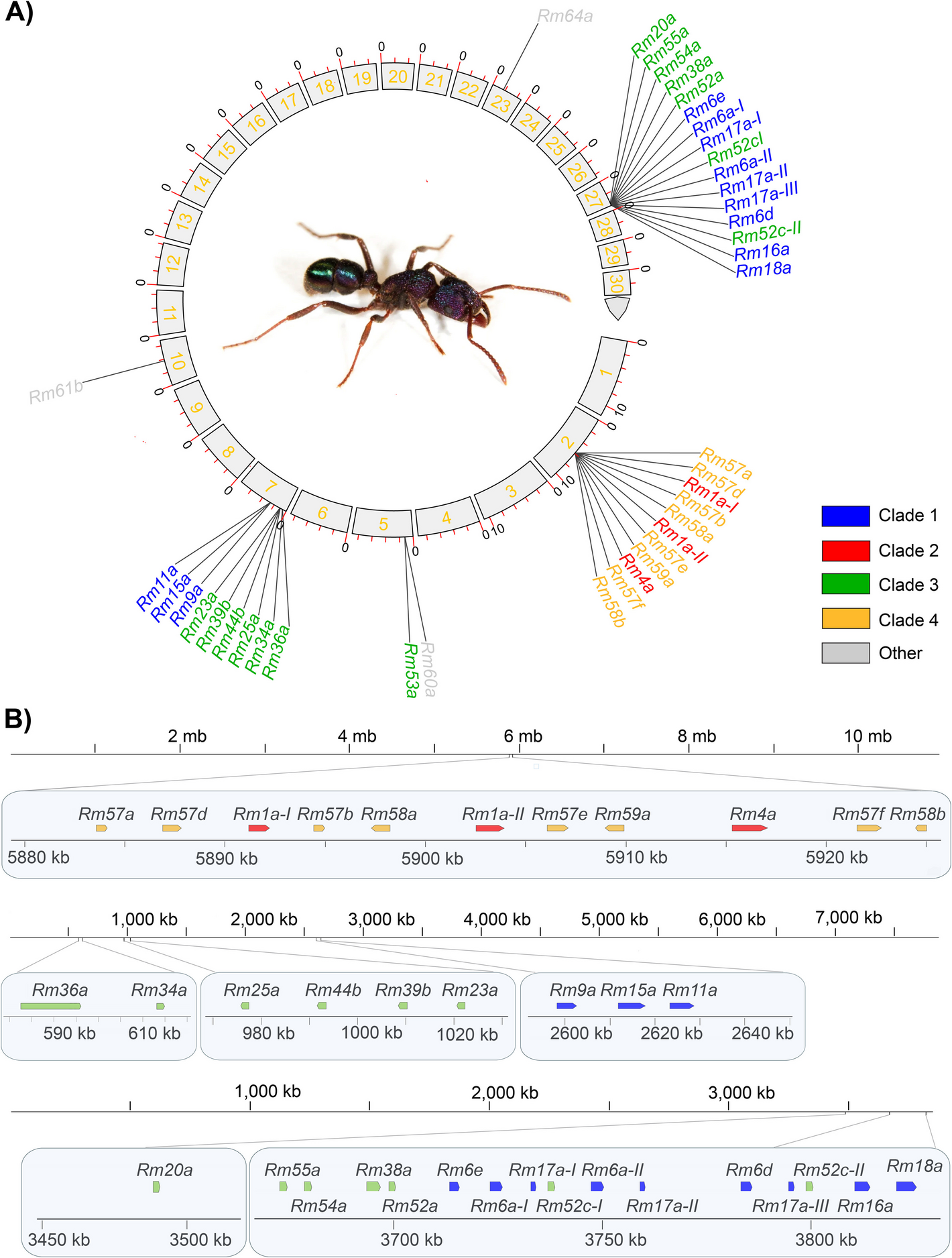
Genome from a single ant resolves allelic and paralogous toxin diversity
Assembling the HiFi reads obtained from DNA isolated from a single worker of R. metallica (Additional file 1: Fig. S1) yielded a primary assembly with a total size of 395 Mb (394,639,337 bp) distributed across 507 contigs, which falls into the normal size range compared to other ant genomes [40] and fits well with the k-mer-based estimate of 372.1 Mb (Additional file 1: Fig. S2A). The assembly has high contiguity, with a maximum contig size of 11 Mb (11,586,707 bp), L50 value of 33 (number of ranked contigs containing half the assembly length), N50 value of ~ 3 Mb (3,430,620 bp), and N90 value of ~ 500 kb (494,692 bp). Genome-assembly completeness is 97.0% as measured against hymenopteran BUSCOs, of which 94.8% are complete single-copy BUSCOs and 2.2% are duplicated BUSCOs (Additional file 1: Fig. S2B). The final mitogenome assembly has a size of 17,121 bp, containing 37 identified genes in total, of which 13 are protein coding genes, 22 tRNA genes, and 2 rRNA genes (Additional file 1: Fig. S3). The mitogenome is distributed onto many different contigs of shorter size (Additional file 2: Table S1). As these did not interfere with the toxin-encoding regions, we did not remove these contigs from the final genome assembly before submission to NCBI.
Annotation using the funannotate pipeline resulted in identification of total of 31,255 genes. Although this overall gene annotation is relatively complete—as determined by measuring against both hymenopteran BUSCOs and non-ectatotoxin venom components—the annotated gene models included only nine ectatotoxin loci: three loci on contig 2 (two identical coding sequence regions of Rm1a and Rm4a), four loci on contig 7, and two loci on contig 27. These results highlight the difficulties in predicting genes encoding hypervariable short peptides such as ectatotoxins or other structurally similar short peptide toxins, which represent the main toxins of ants and other hymenopterans, and in R. metallica constitute almost 99% of the expressed venom [30]. We therefore annotated the 100 ectatotoxins identified in the colony venom gland transcriptome of R. metallica [30] by mapping their amino acid sequences onto the genome with both exonerate and miniprot. In addition, we mapped transcriptome reads from the same study and used ToxCodAn-Genome [41] for extra validation, supplemented by manual curation using FGENESH + (see methods). Using miniprot together with manual curation resulted in the annotation of a total of 45 ectatotoxin loci (Additional file 2: Table S2). The GC content within these ectatotoxin-encoding genes is approximately 46% compared to the overall GC content in the genome, which is 37.7%. Annotation of ectatotoxin genes using ToxCodAn-Genome specifically trained on ectatotoxins returned 32 of these 45 loci, suggesting toxin-specific gene predictors may be useful in identifying genes encoding short and hypervariable peptides.
In addition to ectatotoxins, we also identified several loci with genes encoding non-ectatotoxin venom proteins and peptides such as dipeptidyl peptidase 4 (DPP-4), phospholipase, CAP, crustacean neurohormone (CNH) and several precursory EGF-domain peptides previously described from the venom of R. metallica [30, 42]. Each of these venom components is located on different contigs from the ectatotoxins. The five EGF-like peptides identified from the pooled transcriptome data (ECTX2-Rm1a, ECTX2-Rm1b, ECTX2-Rm1c, ECTX2-Rm1d, ECTX2-Rm1e) are located on contig 4 and are encoded by one locus, suggesting that they are alleles. Dipeptidyl peptidase-4 (DPP-4) is also located at a single locus on contig 4, while the CNH is a single gene with three exons on contig 115. Although they can be detected in the venom, these non-ectatotoxin peptides and proteins are not major components and constitute less than 1.2% of the total expressed venom [30]. Hence, we focused on the ectatotoxins for all the subsequent analyses.
Venom-encoding ectatotoxin diversity is encoded by a few clustered gene regions
Of the identified 45 ectatotoxin-coding loci, 36 are located within five different clustered gene regions distributed on mainly three contigs, namely contig 2, 7, and 27 (Fig. 1A, B): Contig 2 (~ 10.9 Mb) contains one ectatotoxin region in the middle of the contig (bp 5,883,552–5,925,068) that comprises eleven loci. Contig 7 (~ 7.9 Mb length) contains three ectatotoxin clusters, with 2, 4, and 3 loci for each region, respectively. These regions are located at the start of the contig in the forward orientation (total region bp 595,883–2,643,941). Contig 27 (~ 3.85 Mb length) contains one main gene cluster with 15 ectatotoxin-coding loci spanning bp 3,672,679–3,825,124. It also contains one single locus (Rm20a) downstream with location bp 3,488,477–3,490,334 (Fig. 1B). In addition to the 36 loci on contig 2, 7, and 27, additional loci are found on contigs 5 (2 loci), 10 and 23 (1 locus each), 35 (2 loci), 52 (1 locus), and 141 (2 loci). Previously, Robinson et al. [30] grouped the ectatotoxins (there referred to as aculeatoxins) into three different clades based on structural similarities: Rm1a–Rm5b constitute clade 2, Rm6a–Rm19a constitute clade 1, and Rm20a–Rm55b constitute clade 3. Here, we also define an additional fourth clade, which includes the ectatotoxins Rm57a–Rm61c (Additional file 1: Fig. S4).
Toxin-encoding genes in the primary genome assembly of R. metallica. A Circular plot of the primary genome assembly showing the distribution of loci encoding ectatotoxins on the first 30 contigs, which correspond to almost half the assembly length. The components were identified from the pooled secreted venom of R. metallica [30]. B Locations of clustered ectatotoxin-encoding loci on contigs 2 (top), 7 (middle) and 27 (bottom). Contig 2 has one gene cluster containing ectatotoxins from clades 2 and 4. Contig 7 has three clusters, where each contains separate toxins from either clade 1 or clade 4. Contig 27 has one cluster plus one additional locus. The five clustered regions and the single locus on contig 27 contain 36 identified ectatotoxin-coding loci. The Rm36a locus on contig 7 likely represents two pseudogenized loci
The ectatotoxin-encoding genes have two main exon structures. The first type is characterized by two coding exons, in which the signal- and propeptide are coded by the first exon, and the mature peptide is coded by the last part of the first exon plus the entire second exon (Fig. 2A). Type 2 consists of three exons: Like type 1, the signal- and propeptide are coded by the first exon, while the mature part of the exon is coded by the last part of the first exon together with exons two and three (Fig. 2A). Of the 36 clustered ectatotoxin loci, 24 are of type 1—the most common type—while eight loci are of type 2, and four remain uncertain due to pseudogenization. Type 1 is found within all the clusters on all three contigs, while type 2 is mainly located on the largest cluster on contig 27, comprising five loci at contig 27 (Rm20a, Rm38a, Rm52a, Rm52c-I, Rm52c-II) and three loci on contig 7 (Rm34a, Rm44b, Rm39b) (Fig. 2B). Hence, contig 2 exclusively consists of type-1 ectatotoxin genes, while contig 7 and 27 contain both. All the type-2 gene structures are ectatotoxins that belong to clade 3, reflecting their close relatedness. However, on contig 27, all clade-3 ectatotoxin-encoding genes are of type-2 gene structure, except for Rm55a, which is the only clade-3 toxin with a type-1 gene structure annotated to contig 27.

Ectatotoxin-encoding genes comprise two types of structures. A The type-1 ectatotoxin gene structure (top) consists of two exons, with the first containing signal- (black) and propeptide (cyan) as well as the first residues of the mature toxin (orange). In the type-2 ectatotoxin gene structure (bottom), the exon encoding the mature toxin peptide is split by a short intron. The length (81 bp) of the second intron is highly conserved among type-2 ectatotoxin genes, which belong mainly to clade 3 (located on both contig 7 and 27). Black and cyan regions correspond to signal- and propeptide, respectively, while the orange regions correspond to the mature part of the peptides. B Ectatotoxins, their respective clades and gene structures, and their distribution across the three contigs. Contig 2 consists only of type-1 ectatotoxin genes while contig 7 and 27 contain both types
The second intron in type-2 genes is both short (less than 100 nucleotides) and highly conserved with respect to sequence similarity. Five of the eight type-2 genes have second introns with a length of 81 bp, one gene has a second intron of 82 bp, there are two identical coding regions of Rm52b with 90 bp second introns, and Rm39b has a second intron of 98 bp. However, some of the most similar introns belonged to toxin genes that are not in genomic proximity. For example, Rm20a, Rm44b, and Rm34a cluster with respect to their second intron and the lowest pairwise similarity among these is 97.6%, with only two nucleotide substitutions. But while Rm34a and Rm44b are located on contig 7, Rm20a was found as a single locus on contig 27. Another example is Rm52a and Rm38a, which are in close genomic proximity to each other (2510 bp; Fig. 1B): Although their exon-based phylogenetic relationship suggests they are closely related (Fig. 3), their second introns are poorly conserved, with a pairwise identity of only 61.1%. These findings raise interesting questions about the evolution of the different gene structures, as closely related toxins neither necessarily have the same gene structure nor are found in close proximity (see also Fig. 3).
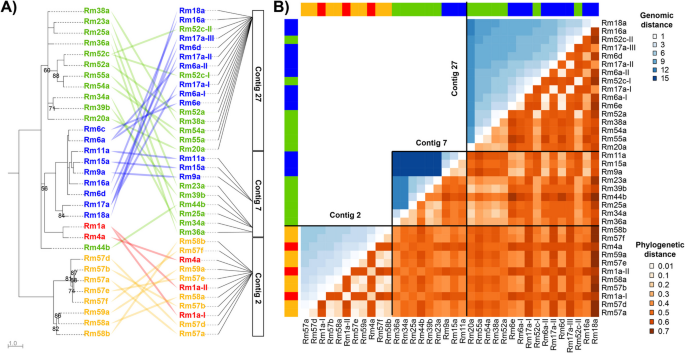
Ectatotoxin loci show a complex gene-family expansion pattern. A Ectatotoxin-locus phylogeny (left) is a poor predictor of physical location in the genome (right). Phylogeny is estimated by maximum likelihood under the VT + G4 model. Nodes with bootstrap support < 50% are collapsed, and only support values < 90% are shown. Colors correspond to clades 1–4 as per Fig. 1. Amino acid sequence alignment is provided as Additional file 3. B Heatmap of physical genomic distance (blue, above diagonal; log2-adjusted bp) and phylogenetic distance (red, below diagonal) illustrating pairwise comparison of all the ectatotoxin loci. Color bars on the left and on top indicate toxin clade (clade 1 = blue, clade 2 = red, clade 3 = green, clade 4 = orange). Contig 2 corresponds to the bottom left blue triangle, contig 7 to the middle blue triangle, and contig 27 to the upper blue triangle to the right. Pairwise comparison of genetic distances within each contig is shown in blue, while pairwise comparison of phylogenetic distances between the 36 ectatotoxin loci is shown in red. Physical genomic distances are measured in base pairs with log-adjusted values, with darker blue indicating greater distance. Phylogenetic distances are calculated as pairwise identities using nucleotide alignment of the mapped loci, with darker red indicating greater genetic distance. Nucleotide sequence alignment and distance tables are provided as Additional file 4 and Additional file 2: Table S3, respectively
Ectatotoxin genes show evidence of active gene-family expansions
Given the tandem repeats of ectatotoxins, we next examined whether dynamic and recent expansions by gene duplication could have contributed to these patterns (Fig. 1B). The ectatotoxin cluster on contig 2 includes two Rm1a-encoding loci with identical coding sequence exons but with different intron lengths (820 bp versus 1230 bp) due to repeated insertions of -GTGTGTGT- and -GTGCGTGC-. Besides these insertions, the introns are highly similar (97.7%), suggesting that the duplication of the locus encoding Rm1a might be a recent evolutionary event. Similarly, the ectatotoxin cluster on contig 27 also contains multiple identical coding sequence loci: Rm6a and Rm52c are encoded by two loci each while Rm17a is encoded by three loci. All the Rm6a- and Rm17a-encoding loci contain one intron (type-1 gene structure). In Rm6a-I and Rm6a-II, the introns are both 2488 bp and identical. Among the three Rm17a-coding loci, Rm17a-I has an intron length of 634 bp, while in Rm17a-II and Rm17a-III the intron length is 630 bp and 632 bp, respectively. Rm17a-I has an inserted -TATA-motif which is not present in Rm17a-II. In Rm17a-III, there is a deletion of -TA- with respect to Rm17a-I. In addition, the introns in Rm17a-II and Rm17a-III have two nucleotide substitutions. The duplicated Rm52c-encoding genes consist of two introns each (type-2 gene structure). The first intron differs in length between Rm52c-I (922 bp) and Rm52c-II (924 bp) and has a pairwise similarity of 96.5%, including several indels and substitutions. The second introns, which are much shorter (90 bp in Rm52c-I and 81 bp in Rm52c-II), have a pairwise similarity of 96.3%. This high similarity between the introns may indicate that expansions by tandem duplication of ectatotoxin genes have played an important, and ongoing, role in their evolution.
Distributions of ectatotoxins show a complex phylogenetic/genomic relationship
To examine the patterns involved in ectatotoxin gene-family expansions in more detail, we compared the phylogenetic relationships of ectatotoxins with their physical distribution in the genome. This approach revealed that several clusters contain ectatotoxins from multiple clades and that their phylogenetic relationships are poor predictors of their genomic distributions (Fig. 3A). Of all clusters of ectatotoxin loci in R. metallica, only the three clustered regions in contig 7 consist of paralogs belonging to the same clades—the first two consist exclusively of toxins from clade 3, while the third region consists exclusively of toxins from clade 1. In contrast, the ectatotoxin cluster on contig 2 contains all toxins in clade 2 and clade 4 but interspersed with each other. Contig 27 also contains regions where two loci encoding clade 3 ectatotoxins are present between nine loci encoding clade 1 ectatotoxins. Thus, the clusters on contig 2 and 27 are mosaics of toxins from different clades with an overlapping tandem arrangement. Indeed, mapping the phylogenetic relationships against the genomic distance within clustered regions and contigs further highlighted several discrepancies between toxin distribution and phylogenetic relationship (Fig. 3B), suggesting complex family expansions have taken place.
Toxin genes are associated with transposable elements that may have facilitated their functional diversification
The peculiar pattern of discrepancy between sequence similarities and physical genomic distances suggests that there are mechanisms that facilitate transpositions of genomic regions to create patterns of tandem duplications. With the hypothesis that repetitive elements might invoke such gene-family expansions, we annotated transposable elements and compared their distributions across the toxin and non-toxin regions of the genome. The repetitive landscape of the R. metallica genome consists mainly of class II DNA elements along with long terminal repeats (LTRs) and long interspersed nuclear elements (LINEs) (Fig. 4A). The repeat landscape also illustrates the accumulation history of transposable elements and suggests that there are many potentially recently active DNA transposons (Fig. 4A). We also found that all toxin regions contain a high density of transposable elements (Additional file 1: Fig. S5), and that the transposable-element density is higher in the toxin regions compared to the mean TE density of similarly sized windows of their respective contigs, particularly for contig 2 and contig 7, but less so for contig 27 (Fig. 4B).
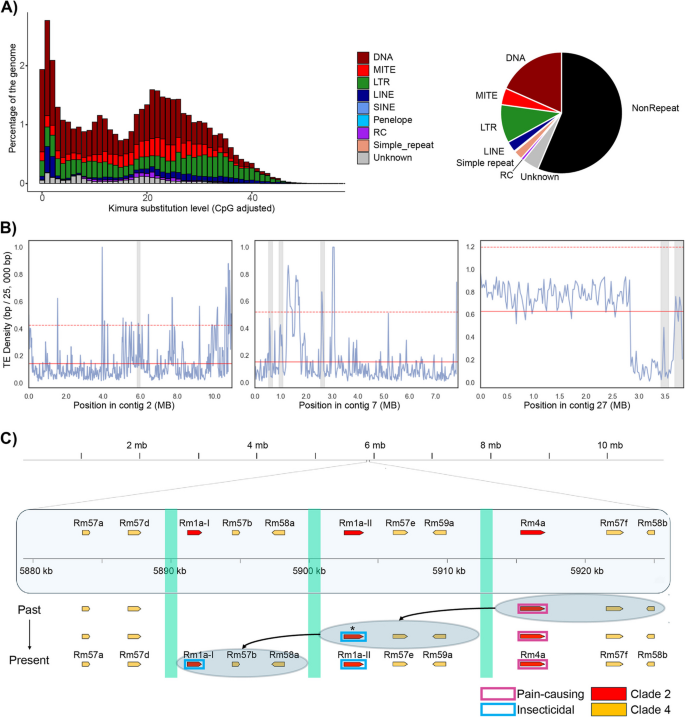
Ectatotoxin gene-family expansions may be facilitated by flanking transposable elements. A Repetitive landscape of the R. metallica genome, showing the amount and relative proportions of the different transposable elements. DNA repeats were the most abundant, followed by LTR, MITE, and LINE. The y-axis refers to the genome coverage for each type of transposable element. The x-axis shows calculated Kimura distances to their corresponding consensus sequence. Bars to the left do not diverge much from the consensus sequence and suggest recent copies, while bars to the right indicate more ancient copies. B Transposable-element density on contig 2 (left), contig 7 (middle), and contig 27 (right) was estimated in windows of 25,000 bp. Grey bars represent the ectatotoxin clusters, the solid red line represents the mean transposable-element density across the region, and the dotted red line represents the mean plus two standard deviations. C On contig 2, the transposable element Tc1/Mariner (turquoise bars) flanks Rm1a and Rm4a, suggesting a mechanistic hypothesis for the evolution of insecticidal Rm1a (blue boxes) derived from the defensive Rm4a (purple boxes). Ectatotoxins of unknown functions belonging to clade 4 are indicated by yellow boxes. The asterisk indicates neofunctionalization
As contig 2 comprises tandem-duplicated regions containing Rm1a- and Rm4a-encoding genes—which are thought to be large-effect loci with respect to pain-causing (defensive) and insecticidal (predatory) activity of the venom [30]—we focused on annotation of the different types of transposable elements surrounding the 11 loci on contig 2 (Additional file 1: Fig. S6–S8). The DNA transposon Tc1/Mariner was annotated to flanking regions of the two tandem-duplicated regions of Rm1a-I, Rm1a-II, and Rm4a (Fig. 4C, Additional file 1: Fig. S6). We also identified a DNA Sola transposon within the Tc1/Mariner region. However, it is likely that the Sola is a “false positive” integrated part of the Tc1/Mariner, and that Tc1/Mariner is the most Likely responsible transposable element for the tandem gene duplicated regions on contig 2 (Additional file 1: Fig. S9–S10). Considering the ectatotoxin phylogenetic relationships [30] and genetic distances (Fig. 3), we hypothesize that Rm1a evolved through a concerted duplication of Rm4a + Rm57f + Rm58b. We consider Rm4a the ancestral state due to the distribution of orthologs in many formicoid ants [43]. Following the functional diversification of the second copies into Rm1a-II + Rm57e + Rm59a, a recent second duplication of these two toxin-encoding genes resulted in a third set of paralogs containing an identical coding-sequence region of Rm1a (Rm1a-I) together with two new toxin-encoding genes Rm57b + Rm58a (Fig. 4C). This hypothesis is also supported by comparing the pairwise identities of Rm57f/Rm57e (97.6%), Rm57e/Rm57b (96.9%), and Rm57f/Rm57b (96.7%). Taken together, these findings suggest that the insecticidal Rm1a evolved from a vertebrate-specific defensive toxin (Rm4a) through gene duplications facilitated by Tc1/Mariner-type transposable elements.
Although Tc1/Mariner seems to be mediating tandem gene duplications of Rm4a + Rm57b + Rm58b on contig 2, similar transposable element annotations did not flank the tandem duplications of the genes encoding Rm17a, Rm6a, and Rm52c on contig 27. Thus, although transposable elements are associated with toxin-coding regions of the genome, we did not identify elements that are universally associated with, or potentially responsible for, all ectatotoxin gene clusters. Further studies are therefore needed to elucidate the evolutionary history and underlying genomic mechanisms of the tandem duplications on contig 27.
Colony-wide venom composition contains functionally distinct ectatotoxin alleles
Although gene duplications explain some of the short venom peptide diversity observed within the colony of R. metallica, the number of ectatotoxin loci still accounted for less than half (45 of 100) of the ectatotoxins previously identified from pooled venom-gland transcriptome data [30]. Indeed, examining the degree of allelic variation in more detail by mapping these ectatotoxins to our primary assembly revealed that most loci have multiple allelic variants. In contig 2, five out of eleven loci (four out of ten non-identical coding regions) have multiple allelic variants, with the previously published pooled venomgland transcriptome (100 ants from the same colony) containing a total of 28 alleles across these loci (mean 4.5 alleles per variable locus). Similarly, the nine clustered ectatotoxin loci on contig 7 account for 35 alleles (mean 4.25 alleles per variable locus), while eight out of the sixteen loci in contig 27 (six out of the twelve non-identical coding regions) have a total of 33 alleles (mean 4.13 alleles per variable locus). Note that this allelic diversity is not restricted to ectatotoxins but is also present in the EGF-domain-containing toxins (ECTX2-Rm1a–e), where five homologs map to a single locus.
In addition to having many allelic variants, there is a striking structural diversity among alleles mapping to each locus. Conducting a pairwise blast analysis on mature ectatotoxin amino acid sequences, we found no patterns of clustering within loci (Additional file 1: Fig. S11). This lack of clustering was also detected when projecting the embeddings of mature ectatotoxin peptides, suggesting substantial functional variation among allelic variants (Additional file 1: Fig. S12). To test the functional implications of this structural allelic variation, we synthesized and tested allelic variants of the two ectatotoxin loci with the clearest known toxicity phenotype. For the insecticidal Rm1a, we synthesized the alleles Rm1b, Rm1c, and Rm2a. For the vertebrate-specific pain-causing Rm4a, we synthesized and tested the alleles Rm3a and Rm5a. We found that 1 µM of Rm1a, Rm1b, Rm1c, Rm2a, and Rm3a all caused an activation of dorsal root ganglion (DRG) cells—which include pain-sensing neurons—defined as an increase in intracellular Ca2+ concentration, although slower and weaker than Rm4a and Rm5a, which activated 96% and 70% of the DRG cells, respectively. Rm1a activated on average 35% of the DRG cells, slightly higher than its corresponding alleles Rm1b (26%), Rm1c (30%) and Rm2a (10%) (Fig. 5A, Additional file 1: Fig. S13). The slow and weak activation of Rm3a (9% activation of DRGs) contrasts with the immediate activation caused by its allelic variants Rm4a and Rm5a (Fig. 5A, Additional file 1: Fig. S13N, Q; see also [30]). Rm5a activated DRG gradually, although slower than Rm4a (Additional file 1: Fig. S13Q, T). Injection into house crickets (Acheta domesticus) revealed that the insecticidal Rm1a and its allelic variants Rm1b, Rm1c, and Rm2a efficiently incapacitated the crickets. Rm1a and Rm1b incapacitated all the house crickets, while Rm1c incapacitated 89% of the house crickets on average (mean, n = 3). Rm2a was substantially less potent than the other three allelic variants, incapacitating 56% of the crickets on average (mean, n = 3). Rm3a, Rm4a, or Rm5a did not have any incapacitating effect (Fig. 5B). These results support the hypothesis that duplication followed by neofunctionalization plays a pivotal role in generating the diverse ectatotoxin arsenal that is present in colonies of R. metallica [30]. Different potency and potentially also function (e.g., Rm3a and Rm2a) further demonstrate that the allelic diversity contributes to an expanded functional toolkit on the colony level.
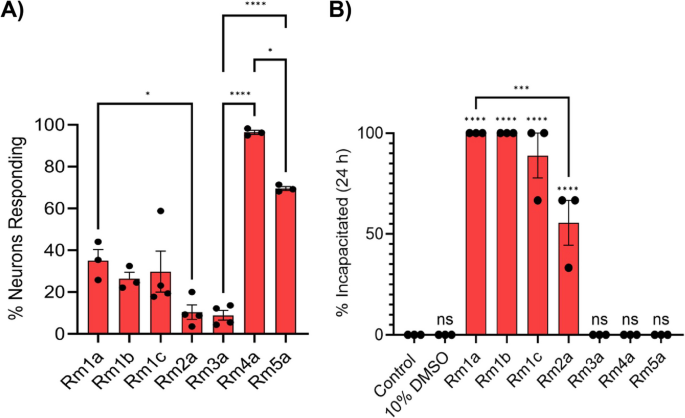
Ectatotoxin alleles exhibit different functional properties. A Effect of R. metallica peptide toxins on mouse dorsal root ganglion cells. Bars show the mean percentage of neurons that responded to 1 µM of each peptide across three independent experiments. Rm1a: mean = 35.1%, CI = 12.4–57.7%; Rm1b: mean = 26.4%, CI = 12.9–39.9%; Rm1c: mean = 29.8%, CI = − 1.2–60.8%; Rm2a: mean = 10.4%, CI = − 0.6–21.4%; Rm3a: mean = 8.9%, CI = 1.5–16.3%; Rm4a: mean = 96.4%, CI = 92.3–100.6%; Rm5a: mean = 70.0%, CI = 65.5–73.6%. CI refers to the 95% confidence interval. B Effect of R. metallica peptide toxins on house crickets (Acheta domesticus). Bars show the mean percentage of crickets incapacitated after intra-abdominal injection of each peptide (1.34 nmol/g; n = 3 independent experiments). Rm1a and Rm1b incapacitated all the crickets, while Rm1c and Rm2a incapacitated 88.9% (CI = 41.1–136.7%) and 55.6% (CI = 7.7–103.4%), respectively. CI refers to the 95% confidence interval. Statistical significance was tested using one-way ANOVA with Tukey’s multiple comparisons test and is indicated in A and B by asterisks: **** indicates p < 0.0001, *** indicates p < 0.001, * indicates p < 0.05. Non-significant p-values are indicated by ns. Significance above columns in B shows comparisons between each treatment and negative control.
Nonsynonymous heterozygous sites indicate elevated selection in toxin regions
Given the combination of recent gene duplications and high intracolony allelic diversity among R. metallica ectatotoxins, we next examined whether this toxin diversity was also reflected in the level and types of heterozygosity. First, we used heterozygous pairwise single nucleotide polymorphisms to compare toxin to non-toxin regions throughout the genome. The heterozygous nucleotide variation per site (1-Kb window) was higher for the regions containing toxin genes, with a mean heterozygosity per site of 1.48% (interquartile range, i.e., Q1–Q3 (IQR) = 0.6–2.2%, median = 1.2%), compared to non-toxic regions in the genome, where the mean heterozygosity was 0.48% (IQR = 0.2–0.6%, median = 0.3%) (Fig. 6A). These results demonstrate that ectatotoxins are genetically diverse compared to the rest of the genome.
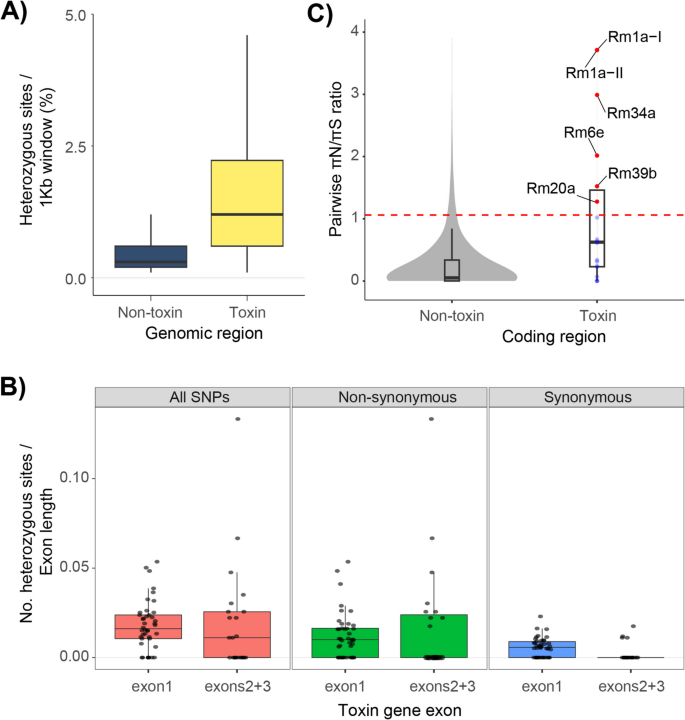
Toxin genes show elevated heterozygosity and high ratio of nonsynonymous heterozygotic nucleotides. A Toxin regions show higher levels of heterozygosity compared to non-toxin regions. Here, heterozygosity is defined as the number of heterozygous nucleotides divided by the total number of nucleotides in each window (toxins mean = 1.475%, non-toxin mean = 0.477%). Boxes show the interquartile range (Q1–Q3) and include the middle 50% of the data, in which the median (Q2) is marked with the horizontal line. B Total, nonsynonymous, and synonymous SNPs among toxin alleles in first exon (signal- and propeptide, n = 135) versus second and third exons (mature part of toxin, n = 77). First exon estimates also include loci of partially annotated genes where only the first exon was identified. Boxes show the interquartile range (Q1–Q3) and include the middle 50% of the data, in which the median (Q2) is marked with the horizontal line. C Pairwise πN/πS ratio of heterozygote nucleotides in toxin and non-toxin coding genes with non-identical alleles. The red dashed Line represents the 95th percentile (= 1.0751) of πN/πS distribution of non-toxin genes. Toxin genes exceeding the 95th percentile of the non-toxin genes are marked with red dots. A mean πN/πS ≈1 for toxin genes fits the neutral prediction, while the mean πN/πS of 0.248 for non-toxin genes indicates purifying selection
We further identified nonsynonymous and synonymous heterozygous nucleotides among the ectatotoxin exons. As the more conserved signal- and propeptide regions are coded by the first exon and the mature peptide is coded by the second and third exons, we expected to find a higher degree of nonsynonymous nucleotides in the second and third exons. Indeed, although we found a high number of nonsynonymous heterozygotic nucleotides at all exons, the second and third exons had a higher number of nonsynonymous heterozygotic nucleotides (IQR = 0–0.024 nucleotides per exon length) compared to the first exon (IQR = 0–0.016 nucleotides per exon length) (Fig. 6B). There were also more synonymous heterozygous sites on the first exon (IQR = 0–0.009 nucleotides per exon length) compared to the second and third exons, which have few synonymous heterozygotic nucleotides. This is what we would expect from conserved genic regions.
Finally, we estimated the rates of nonsynonymous to synonymous heterozygous sites (πN/πS) from pairwise comparisons of reference and alternate single-nucleotide polymorphisms. This approach revealed higher πN/πS ratios among toxin regions compared to non-toxin coding regions (toxin mean πN/πS = 0.95, IQR = 0.23–1.46, n = 48; non-toxin mean πN/πS = 0.25, IQR = 0.001–0.34, n = 9407) (Fig. 6C). Thus, while regions containing toxin genes have higher than expected heterozygosity and elevated nucleotide variation compared to the rest of the genome of R. metallica, a πN/πS ≈ 1 is a close fit to the neutral prediction. However, many of the ectatotoxins also have πN/πS > 1, such as Rm1a and its alleles, as well as Rm34a, Rm6e, and Rm20a, among others, suggesting these may be under strong positive selection (see Fig. 6C).