Calculating the excited states of molecules presents a significant challenge for quantum computing, and researchers continually seek methods to improve the accuracy of these calculations on current-era devices. Bikrant Bhattacharyya from the California Institute of Technology, Gokul Ravi from the University of Michigan, and colleagues have developed a new approach, termed Excited-CAFQA, to initialise calculations of molecular excited states. Building on previous work that successfully optimised ground state calculations, this method performs a classical search through a limited set of states to find ideal starting parameters for a quantum algorithm. The team demonstrates that Excited-CAFQA achieves remarkably high accuracy, consistently reaching 90 to 99% across various molecular systems and excited states, representing a substantial step towards reliable quantum simulations of molecular behaviour.
is often insufficient for quantum chemistry applications. One method of improving Variational Quantum Algorithms (VQAs) is through accurate ansatz initialization. The Clifford Ansatz For Quantum Accuracy (CAFQA) protocol performs a discrete search through a classically simulatable subset of the entire state space to find a desirable initialization. Prior work has evaluated CAFQA applied to the Variational Quantum Eigensolver (VQE), a VQA that computes ground states of a Hamiltonian. Motivated by CAFQA’s success, scientists propose Excited-CAFQA initialization for Variational Quantum Deflation (VQD), a quantum algorithm that extends VQE by allowing the computation of excited states. VQD recursively computes excited states by constraining.
CAFQA Optimizes VQE Initial Parameters Reliably
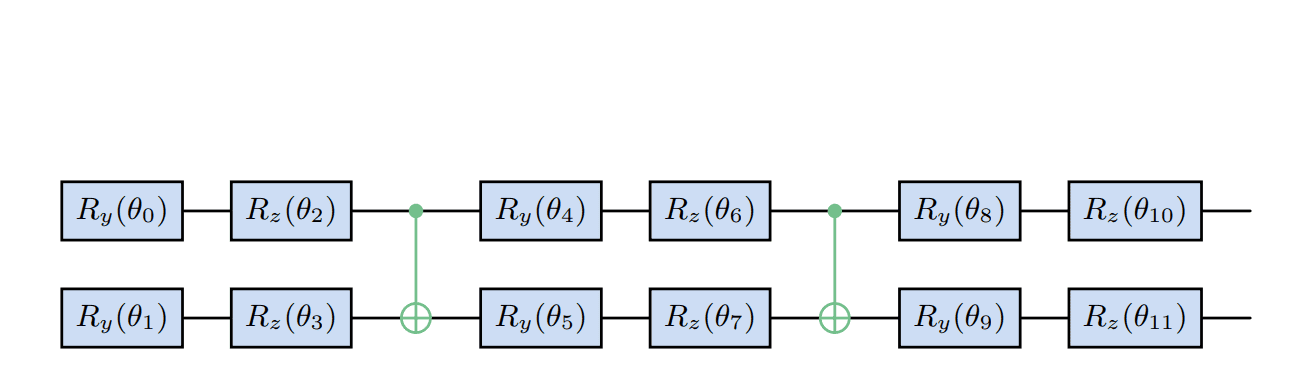
The Variational Quantum Eigensolver (VQE) relies on the Variational Principle, which states that the ground state energy is minimized by a specific quantum state. To find this ground state, VQE parameterizes a quantum state and then iteratively minimizes the energy expectation value using a classical optimizer, repeatedly querying a quantum device to evaluate the energy and update the parameters. The performance of VQE can be limited by noise in near-term quantum devices, making accurate initial parameters for the ansatz crucial. The Clifford Ansatz For Quantum Accuracy (CAFQA) protocol addresses this by performing a classically simulatable search over a discrete parameter space to find appropriate initial values before beginning the continuous parameter optimization loop.
CAFQA restricts the search to parameter values that result in Clifford circuits, which can be efficiently simulated classically. To extend this approach to finding multiple excited states, Variational Quantum Deflation (VQD) is employed. VQD aims to find the first k eigenstates of a Hamiltonian by iteratively finding the ground state, then constructing a new cost function that constrains subsequent optimizations to produce states orthogonal to previously found eigenstates. This process is repeated recursively to find higher excited states. To incorporate Clifford initialization into VQD, the team defined a state that minimizes the expectation value over all Clifford states, and then defined corresponding Clifford loss functions.
These optimizations can be carried out classically because the inner product of stabilizer states can be computed efficiently using their corresponding stabilizer groups. The team demonstrated this approach, termed CAFQA-VQD, on H2 and HeH+ molecules, both of which correspond to 2-qubit Hamiltonians. The results show that the Clifford approximations for the energies converge to the exact values at both small and large bond lengths, with the maximum error occurring near the equilibrium bond length. The optimizer used was a black-box Bayesian optimizer, and parameter transfer was employed, using the optimal parameters from one bond length as the initial point for the next. This approach effectively combines classical pre-optimization with a variational quantum algorithm to improve the accuracy and efficiency of excited state calculations.
Excited-CAFQA Boosts Excited State Energy Accuracy
Scientists have developed Excited-CAFQA, a new initialization protocol for variational quantum deflation (VQD) algorithms, significantly improving their accuracy in calculating excited state energies. Building upon the success of the Clifford Ansatz For Accuracy (CAFQA) method used in variational quantum eigensolver (VQE) calculations, this work extends the approach to determine excited states, a crucial step for understanding molecular properties and chemical reactions. The VQD algorithm recursively computes excited states by constraining each new state to be orthogonal to previously calculated ones, a process enhanced by the accurate initialization provided by Excited-CAFQA. Experiments demonstrate that Excited-CAFQA achieves remarkably high accuracy, ranging from 90 to 99%, across a variety of bond lengths and excited states for both hydrogen (H2) and helium hydride (HeH+) molecular systems.
The maximum error observed for H2 was 0. 087 Ha, while for HeH+ it reached 0. 097 Ha, demonstrating consistent performance across different molecular structures. Results show that the Clifford approximations converge to the exact energies at both small and large bond lengths, with the largest discrepancies occurring near the equilibrium bond length where energy changes are most sensitive. This breakthrough utilizes a classical optimization process to identify optimal initial parameters for each energy level calculation, leveraging the fact that the cost function can be efficiently computed classically within the discrete CAFQA search space. Furthermore, the team implemented parameter transfer, utilizing the optimal parameters found for one bond length as a starting point for the next, accelerating the optimization process. This combination of accurate initialization and efficient optimization delivers a substantial improvement in the reliability and speed of VQD calculations, paving the way for more accurate simulations of complex molecular systems.
Excited State Energies via Classical-Quantum Initialization
Researchers have developed a new initialization protocol, termed Excited-CAFQA, to improve the accuracy of variational quantum algorithms, specifically for calculating excited state energies of molecules. Building upon previous work with the Clifford Ansatz For Accuracy (CAFQA) method, which successfully initializes parameters for ground state calculations, this new approach extends the technique to determine energies of higher energy states. The method involves a classical pre-optimization step, searching a limited, classically simulatable space of possible initial parameters to identify those that yield accurate results when used in a quantum computation. Evaluations on hydrogen and helium hydride molecules demonstrate that Excited-CAFQA achieves high accuracy, with errors ranging from 90 to 99% across various bond lengths and excited states.
Importantly, the optimized parameters converge towards the correct values both at short and long bond distances, with the largest errors occurring near the equilibrium bond length. The team also incorporated a parameter transfer technique, leveraging optimal parameters found for one bond length as a starting point for optimizing another, further enhancing efficiency. The authors acknowledge that the current implementation is limited to two-qubit systems, due to the computational cost of classical pre-optimization. Future work will focus on extending the method to larger molecules and exploring strategies to reduce the computational demands of the classical optimization step. This research represents a significant step towards developing more reliable and accurate variational quantum algorithms for simulating molecular systems and understanding their properties.
👉 More information
🗞 Excited-CAFQA: A classical simulation bootstrap for the variational estimation of molecular excited states
🧠 ArXiv: https://arxiv.org/abs/2509.20588