SV landscape in farmed European seabass
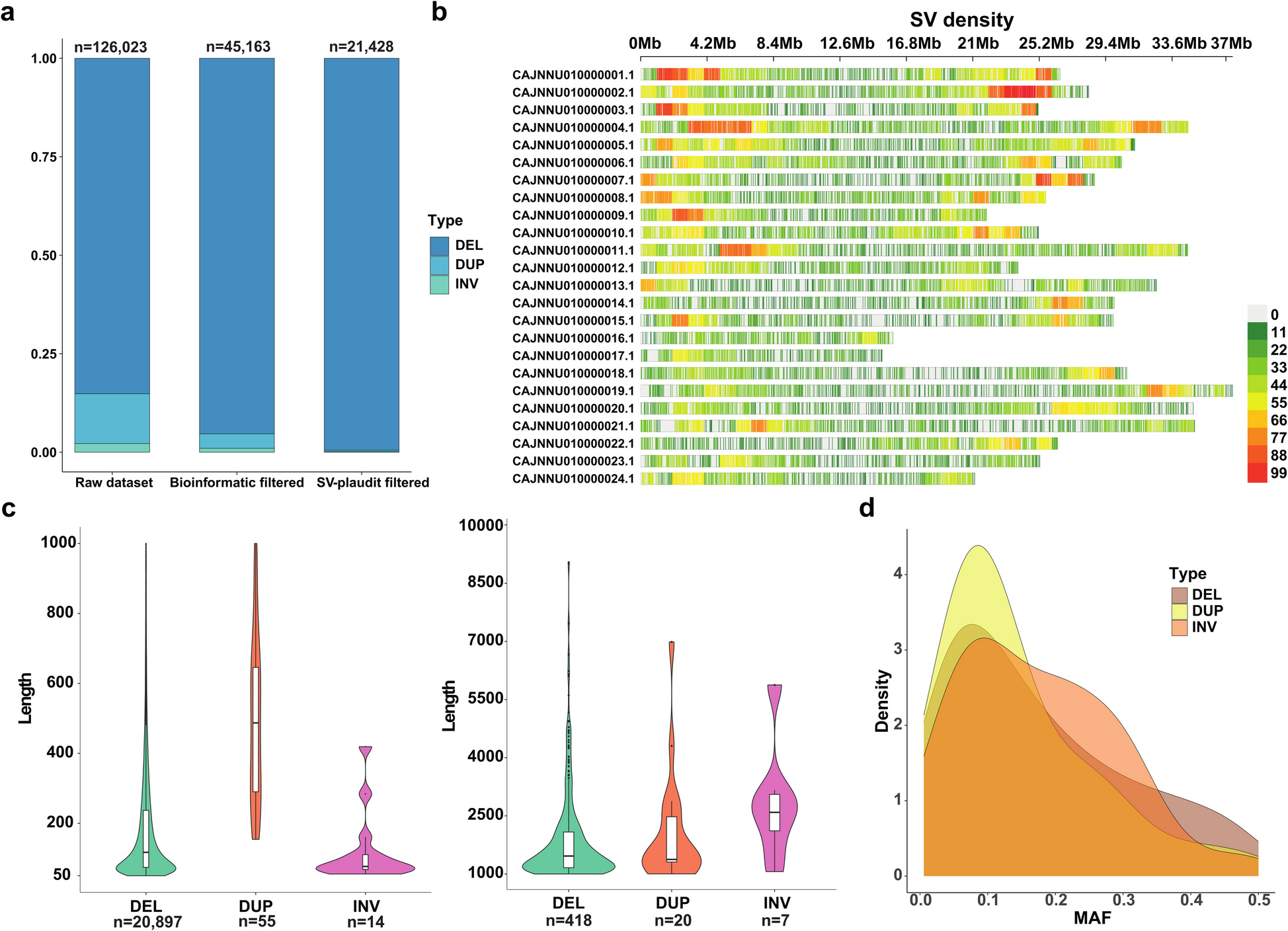
126,023 SVs were initially identified from n = 90 whole genome sequencing (WGS) samples representing a farmed group of European seabass. After filtering of raw SV calls, 45,163 unique SVs were identified, representing 43,057 deletions, 1,654 duplications and 452 inversions (Fig. 1a). Using SV-plaudit, this dataset was reduced to 21,428 high-confidence SVs, representing 21,320 deletions, 75 duplications, and 33 inversions (Fig. 1a). Information about SV size, genomic locations, allele frequencies and SnpEff annotations are provided in Additional file 1: Table 1. We further generated a table of putative orthologous genes for every European seabass gene to facilitate subsequent data interpretations (Additional file 2: Table 2).
High-confidence SVs mapped across the European seabass genome. The data represents 21,248 SVs from a farmed population of n = 90 individuals. a SV counts before and after filtering steps. b SV density per 1 Mb intervals along different chromosomes. c Violin plot for deletions (DEL), duplications (DUP) and inversions (INV), split into two length ranges: 50 to 1,000 bp and 1,000 to 10,000 bp. d Minor allele frequency (MAF) density distribution plot for each SV class
Across the genome, one SV was detected per 31,394 bp on average, with notable variation in SV density within and across chromosomes (Fig. 1b; Additional file 3: Table 3). The genomic location of regions showing the highest (> 80 SVs per Mb) and lowest (< 10 SVs per Mb) density of SVs are provided in Additional file 4: Table 4. The total genomic length of SVs was 6,510,004 bp, representing 0.94% of the reference genome length. The cumulative length of deletions, duplications and inversions was 4,735,175 bp, 67,711 bp and 1,707,118 bp, respectively. Around 90% of the SVs were < 1.5 kb, with average/median lengths of 241/121 bp (Fig. 1c). Deletions, duplications, and inversions showed average/median lengths of 223/120 bp, 904/619 bp, and 51,732/2,456 bp, respectively. The largest deletion, duplication and inversion was 32,027 bp, 6,987 bp and 402,664 bp, respectively, while 16 (0.08%), 1 (1.3%), and 13 (39%) variants from each SV category exceeded 5,000 bp (Additional file 1: Table 1). Three very large inversions (each ~ 400,000 bp) were co-located on CAJNNU010000016, showing > 95% overlap (Additional file 1: Table 1). These inversions have slightly different breakpoints and likely represent the same or closely related variants validated independently using SV-plaudit. The minor allele frequency (MAF) distribution of different SV types showed a peak of around 0.1 in each case (Fig. 1d) and 4,026 SVs exhibited MAFs < 0.05. The number and size distribution of SVs was similar to that observed in previous studies of salmonids using a similar pipeline [19, 21].
SV annotation and functional impacts
SnpEff was used to annotate the 21,428 high-confidence SVs in the European seabass genome (Fig. 2a; Additional file 1: Table 1). Approximately 39%, 25% and 27% of all SVs were located within introns, intergenic regions, and 5,000 bp up/downstream of genes, respectively (Fig. 2b). A small fraction (3.19%) were located within untranslated regions (UTRs). Another small fraction (2.31%, 494 out of 21,428) were categorized as ‘high impact’ by SnpEff, representing 1.83% of all predicted SV impacts (Fig. 2a-b). An UpSet plot was used to visualize relationships between predicted impacts of all SVs, inclusive of high impact effects, which included exon losses, frameshift mutations, and gene fusions (Fig. 2b). A caveat is that the predicted SnpEff impacts depend on the accuracy of genome annotation—hence, while these results provide a valid global overview of SV effects, the impacts of SVs on specific genes warrants careful inspection of the quality of gene model predictions.
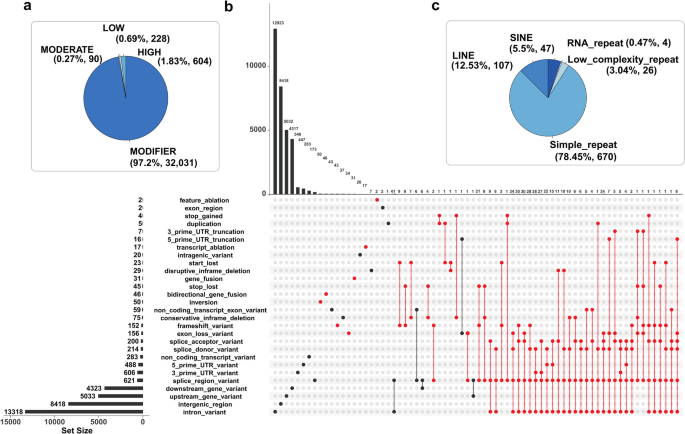
Annotation of SVs in the European seabass genome. The data represents 21,248 SVs from a farmed population of n = 90 individuals. a Proportions of SnpEff categories across the full dataset. b UpSet plot visualising the number of shared SnpEff annotations across different predicted impact categories. Each SV can have multiple SnpEff annotations, with varying impacts. High impact categories were indicated by red dots. c Number and type of repeat elements showing > 50% reciprocal overlap with SVs across the full dataset
Association between SVs and repeats
Past work using the same SV detection pipeline identified recently active repeat elements in the Atlantic salmon genome [19]. The same study identified a large number of deletion SVs of near identical length, representing closely-related sequences belonging to an evolutionarily young transposable element (TE) family specific to Salmo [19]. To explore the relationship between SVs and repetitive sequences and seek evidence for recently active TEs in the current study, we predicted repeat sequences and overlapped them with the 21,428 SVs from farmed seabass (Fig. 2c; Additional file 5: Table 5). 854 SVs (~ 4%) showed a > 50% reciprocal overlap with individual repeat elements, representing 670 simple repeats, 107 long interspersed nuclear elements (LINEs) and 47 short interspersed nuclear elements (SINEs) (Fig. 2c; Additional file 5: Table 5). Among these, only 73 and 6 reciprocal overlaps exceeded 90% and 99%, respectively.
Consequently, there is little support for a strong relationship between SVs and repeat sequences in the European seabass genome. Moreover, our SV dataset captures little evidence for recently active TEs, though it should be noted the SV detection pipeline deployed was not designed for this purpose. This finding sits in contrast to past studies in Atlantic salmon and rainbow trout, where a markedly larger overlap between high-confidence SVs (detected using a similar pipeline) and TEs was reported [19, 21]. For instance, Liu et al. (2021) showed that the repeat sequence content of rainbow trout SVs exceeded 85% [21]. Understanding the basis for such strikingly distinct relationships between SVs and repeat elements across teleost lineages will require further datasets, spanning more species.
Are genes affected by high-impact SVs functionally enriched?
Given the potential of SVs to disrupt gene functions, we tested whether specific biological processes or pathways are enriched among the 487 genes affected by high impact SVs according to SnpEff (Fig. 3; Additional file 6: Table 6 and Additional file 7: Table 7). We identified 233 enriched GO terms and 21 enriched KEGG pathways at uncorrected p < 0.05, but none remained significant after correcting for multiple comparisons (Fig. 3a, b). Terms enriched at p < 0.05 were explained by 285 unique genes (264 genes for GO; 90 genes for KEGG). This overall lack of strong enrichment suggests that SVs with predicted high impacts are not strongly biased towards particular biological functions. The most enriched KEGG pathways (all uncorrected p < 0.01) hinted at weak enrichment of high-impact SVs in immune genes associated with antibacterial functions, including “Yersinia infection”, “C-type lectin receptor signaling pathway” and “Pathogenic Escherichia coli infection”.
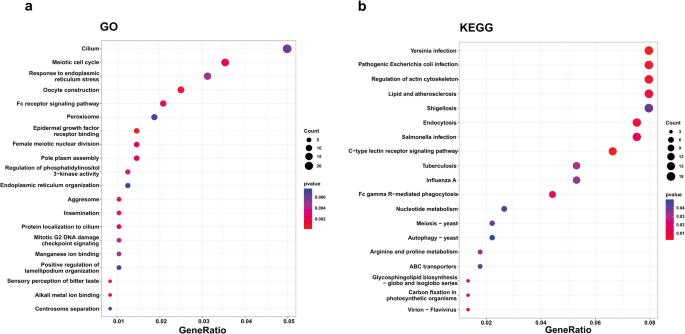
Enrichment tests for genes associated with high impact SVs in the European seabass genome. The analysis was performed using SVs from a farmed population of n = 90 individuals. Clusterprofiler plots are shown for a GO terms and b KEGG pathways. The x axes show the ratio of high impact SV affected genes in each term relative to the total number of SV related genes in that term. Dot sizes show the number of high impact SV affected genes per term, while color is the uncorrected p value. Full results are provided in Additional files Tables 6–8
While not significant following correction, these enrichment tests provide a useful approach to group candidate genes affected by high-impact SVs based on shared functions (provided in Additional file 8: Table 8). For example, with respect to KEGG pathway “C-type lectin receptor signaling pathway”, we identified a 4,018 bp deletion (MAF: 0.09) that removes four out of five coding exons of a C-type lectin domain encoding gene (ENSDLAG00005002384), predicted to disable its coding potential (Additional file 8: Table 8). In the same pathway, a high impact 1,594 bp deletion (MAF: 0.12) is predicted to delete two exons encoding a large segment of another C-type lectin protein domain in a separate gene (ENSDLAG00005017891) predicted to represent a distant homolog of human CLECF4.
Are genes affected by high impact SVs enriched for conserved protein domains?
As an alternative approach to investigate the impact of SVs on functions encoded within the European seabass genome, we asked if proteins encoded by genes affected by high impact SVs according to SnpEff are enriched for particular conserved functional domains. The proportion of InterPro domain annotations associated with the 487 genes affected by high impact SVs was compared to the remaining genes not affected by high impact SVs according to SnpEff (22,482 genes) using Fisher’s Exact tests (Additional file 9: Table 9). 27 InterPro domains showed enrichment at uncorrected p < 0.01, explained by 75 unique coding genes. After correcting for multiple comparisons, only 2 InterPro domains remained significantly enriched at adj. p < 0.05, explained by 4 unique coding genes (Additional file 9: Table 9). However, as Fisher’s Exact test is conservative, we accepted protein domains showing enrichment at uncorrected p < 0.01 to avoid false negatives.
The most enriched protein domains were “Transposase, L1” and “L1 transposable element, C-terminal domain” (p = 1.31e-05 and 3.73e-05, fold enrichment = 46.2 and 138.5, respectively), explained by four transposon coding genes (three shared by both InterPro domains). L1 transposons, also known as long interspersed nuclear element-1 (LINE-1), are extremely common in mammalian genomes, where they are the only known autonomously active retrotransposon, with almost all inactivated by mutations and SVs [34]. In the European seabass genome, each of the four genes encoding these LINE-1 associated domains (ENSDLAG00005000995, ENSDLAG00005026482, ENSDLAG00005030046 and ENSDLAG00005031661) have deletions disruptive to coding regions that are present at moderate to high allele frequencies (respective MAFs: 0.37, 0.37, 0.13, 0.35). Orthologs for several of these LINE-1 genes are predicted in other teleost species by Ensembl, indicating ancient retrotransposition dates. The enrichment of disruptive SVs in genes encoding L1 transposon domains, taken in light of the small number of genes containing these domains (8 for “L1 transposable element, C-terminal domain”; Additional file 9: Table 9) indicates an ongoing process of LINE-1 inactivation.
Another notable enriched domain was ‘5-Hydroxytryptamine 4 receptor’ (p = 4.49e-04, fold enrichment = infinite), which in humans is restricted to a single protein of the same name (abbreviated 5-HT4 receptor, encoded by HTR4), representing a serotonin receptor with essential roles in behaviour, cognition, and gastrointestinal motility and digestion [35]. In seabass, we identified two paralogs related to HTR4 (ENSDLAG00005005527 and ENSDLAG00005011448), predicted by Ensembl to date back to early vertebrates. Both were affected by high impact SVs, representing the only two proteins in the genome harboring the 5-HT4 receptor domain (Additional file 9: Table 9). Another enriched domain was “Alpha-2-macroglobulin, TED domain” (p = 0.0063, fold enrichment = 23.1), explained by two genes (ENSDLAG00005004695 and ENSDLAG00005017199) encoding predicted homologs of Alpha-2-macroglobulin, a family that acts as broad-spectrum protease inhibitors with functions in humoral innate immunity [36]. A large deletion was identified in ENSDLAG00005004695 (MAF: 0.04) that ablates the last 14 exons of this gene (comprising 29 exons).
Other domains showing evidence for enrichment among coding genes affected by high impact SVs have diverse biological roles, including in neurotransmission, myelin formation, intracellular signaling and innate immunity, e.g., ‘Vav, PH domain’, ‘Guanine-nucleotide dissociation stimulator, CDC24, conserved site’, ‘Myelin and lymphocyte (MAL) protein’ and ‘Serine/threonine-protein kinase, active site’, as well as multiple NACHT associated domains. The enriched domain ‘Mitotic spindle checkpoint protein Bub1/Mad3’, is involved in the cell cycle, while ‘ABC transporter type 1’ and ‘Glycosyl transferase family 6’ domains have roles in metabolism, including membrane transport, glycosylation, and nucleotide processing. Domains related to transcriptional and translational regulation, including RNA-binding motifs and kinase domains (e.g., ‘TIAR RNA recognition motif’ and ‘MHCK/EF2 kinase’), highlight possible impacts on gene expression and protein synthesis. Finally, a set of domains such as ‘Somatomedin B’ and ‘Coagulation factor 5/8 C-terminal domain’ were implicated in extracellular matrix integrity and haemostasis.
Overall, this analysis underscores the broad and potentially pleiotropic effects of high-impact SVs on essential cellular and physiological processes in European seabass.
Hundreds of SVs disrupt evolutionarily conserved regions
As a third strategy to explore the impact of SVs on functional features in the European seabass genome, we focussed on genomic regions showing evidence of strong evolutionary constraint across species, a hallmark of purifying selection and functional importance. We overlapped the 21,248 high-confidence SVs from farmed seabass with evolutionarily constrained regions defined from a whole genome alignment of 65 actinopterygian species including European seabass (Fig. 4; Additional file 10: Table 10). These regions were defined using Genomic Evolutionary Rate Profiling, a statistical method that identifies aligned regions showing fewer substitutions than neutral expectations [37].
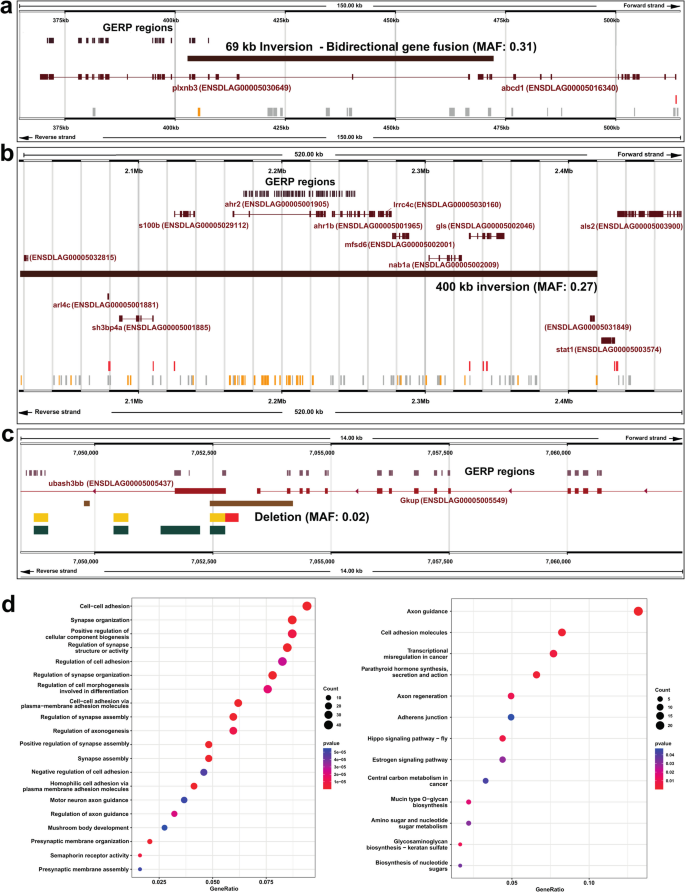
Overlap between SVs and evolutionarily conserved regions in the European seabass genome. The analysis was performed using SVs from a farmed population of n = 90 individuals. a-c Examples of SVs that affect GERP regions in genomic regions containing highly conserved genes discussed in the main text. D Clusterprofiler plots showing the top twenty most enriched GO terms (left) and KEGG pathway (right), using the full set of genes associated with SVs that overlap GERP regions. For KEGG, only the most enriched term “Axon guidance” was significant at adjusted p < 0.05. Full results are provided in Additional files Tables 12 and 13, including tests performed when SVs > 10,000 bp were excluded
366 SVs (1.7%) overlapped 1,219 GERP-defined constrained regions (mean/standard deviation length: 61/77 bp) by at least 1 bp (mean/standard deviation of overlap: 47/58 bp). 17 of these 366 SVs (4.6%) were predicted by SnpEff to represent high impact variants, affecting 40 unique genes. This is approximately twice as high as the background rate of predicted high impact SVs (2.31%; 494 out of 21,428), which is significant (Chi-square test, p = 0.0078). Among these SVs, we identified a 69,461 bp inversion (MAF: 0.31) predicted to cause a bidirectional fusion between the first 7 exons of plxnb3, which overlaps a large cluster of constrained elements, and the last 3 exons of abcd1 (Fig. 4a). plxnb and abcd1 are highly conserved genes, with the European seabass genes predicted to share 51.3% and 67.4% protein-level identity with their human ortholog. In humans, abcd1 encodes ALD, a protein that causes a disease affecting the nervous and adrenal system called adrenoleukodystrophy [38]. plxnb encodes Plexin 3B, a protein central to nervous system development, which acts as a receptor for semaphorin proteins [39]. A remarkably similar mutation involving a deletion spanning abcd1 and plxnb3 is associated with human adrenoleukodystrophy, a rare genetic disease affecting the nervous system and adrenal glands [40].
The three closely-related ~ 400 Kb inversions (MAFs: 0.17–0.27) noted earlier to represent the same variant, in addition to fully-encompassing ten protein coding genes, overlap a cluster of conserved GERP regions located throughout the ahr2 and ahr1b genes (encoding aryl hydrocarbon receptor; AHR, family members), largely affecting non-coding intronic regions (Fig. 4b). Zebrafish orthologs of these genes have roles in xenobiotic metabolism, while ahr2 was required for normal zebrafish behaviour [41]. A group of conserved genes affected by this inversion encode proteins with functions in the brain and nervous system: e.g. s100b, encoding a Ca2 + -binding protein up-regulated in mammalian astrocytes and a key biomarker for multiple nervous system disorders and traumas [42]; mfsd6, encoding a major facilitator superfamily domain-containing protein localised in the mouse brain, with a potential role in energy homeostasis [43]; gls, encoding glutaminase A, an enzyme essential for brain homeostasis that catalyses glutamine hydrolysis to produce glutamate—an important brain excitatory neurotransmitter – that has been associated with many neurodegenerative diseases [44]; arl4c, encoding a small GTPase important for mammalian brain development [45]; and lrrc4c, encoding a leucine-rich repeat protein with key roles in nervous system development [46]. Even genes proximal to this inversion have roles in the nervous system, including stat1, a master immune transcription factor with key roles in regulating brain inflammation and neural stem cell renewal in mice [47, 48], and als2, encoding a rho guanine nucleotide exchange factor called Alsin, which is highly expressed in neurons and the causal gene for characterised neural sclerosis diseases in humans including AMS [49].
Among the high-impact SVs overlapping conserved regions, we identified a 1,761 bp deletion (MAF: 0.02) predicted to ablate the last two coding exons of a gene called gkup (ENSDLAG00005005549; encoding glucuronokinase), which is a highly conserved single copy gene found across teleosts (e.g., ~ 80% protein-level identity between zebrafish and European seabass), with the deletion fully encompassing an exon representing a GERP region (Fig. 4c). This deletion also ablates the entire upstream region (including a promoter and enhancer predicted by Ensembl) and overlaps the first coding exon of ubash3bb (ENSDLAG00005005437; encoding ubiquitin-associated and SH3 domain-containing B), another highly conserved vertebrate gene (e.g., ~ 81% protein-level identity between zebrafish and European seabass). The remaining high-impact SVs overlapping conserved/GERP regions are characterised in Additional file 11: Table 11.
We also identified high-confidence SVs not in the high impact SnpEff subset, overlapping evolutionarily conserved non-coding regions, including intergenic regions, which may represent regulatory elements. For example, we identified a 3,409 bp deletion (location—CAJNNU010000002.1:10,854,297–10857705; MAF: 0.03) residing within a large intergenic region containing a large number of GERP elements, with the nearest genes ~ 92 Kb upstream (ENSDLAG00005013971) and ~ 33 Kb downstream (ENSDLAG00005013980). The region overlapped by this deletion is annotated by Ensembl as open chromatin, indicating its functional importance. As another example, we identified a 6,154 bp deletion (location—CAJNNU010000010.1:13,793,442–13,799,595; MAF: 0.07) embedded within a large intron of a gene (ENSDLAG00005024298) encoding SH2 domain containing 3Ca, a highly conserved (e.g., ~ 77% ID between seabass and zebrafish orthologs) adapter protein that influences cell signalling and adhesion. This deletion region includes a large number of GERP elements and four enhancers predicted by Ensembl to have activity in brain and embryos.
The above examples provide evidence that a subset of segregating SVs in European seabass genomes disrupt highly conserved vertebrate genes and non-coding elements, with impacts Likely affecting both protein function and gene regulation. To gain a global overview of the genes affected by these SVs, we performed GO and KEGG pathway enrichment tests on the 496 genes associated with all high-confidence SVs overlapping GERP regions (Fig. 4d; Additional file 12: Table 12 and Additional file 13: Table 13). As very large SVs may overlap a large number of genes, such as the above highlighted inversion example, we also performed the test excluding SVs defined as > 10,000 bp (n = 482 genes). The results were congruent in both tests, and highlighted enrichment of GO terms associated with brain and nervous system functions linked to synapse organisation, structure and assembly, semaphorin receptor activity, regulation of axon guidance, axonogenesis, neuron recognition, forebrain cell migration, ephrin receptor activity, among others (Additional file 12: Table 12). The KEGG analyses identified one term that remained significant after correction for multiple comparisons, “Axon Guidance”, consistent with the GO analysis (Additional file 13: Table 13). Beyond this clear signal of nervous system related functions, we also identified enrichment of terms associated with the development of other organs and systems, including the heart, the urogenital system, the exocrine system, as well as terms associated with embryonic development (Additional file 12: Table 12). These results indicate that genes showing the strongest conservation across teleost evolution are involved in brain functions and development, while further highlighting that SVs segregating in the studied farmed seabass population are likely to have substantial impacts on phenotypic variation, which could add value to the accuracy of genomic selection or ability to detect QTLs in ongoing aquaculture breeding efforts.
A potential role for SVs in European seabass aquaculture domestication
Our discovery of SVs affecting conserved regions of European seabass genes with neurological and developmental functions motivated us to explore the role of SVs in aquaculture domestication. Bertolotti et al. (2020) [19] identified a strong enrichment for brain-expressed synaptic protein coding genes overlapped by SVs showing significantly different allele frequencies between farmed and wild Atlantic salmon, which were suggested to have promoted behavioural changes during domestication. Many of these SV alleles showed increases in frequency in farmed fish, consistent with a scenario where SVs deleterious in wild fish evolved under relaxed purifying or positive selection in farmed Atlantic salmon, owing to differences in rearing environment [19].
To explore the role of SVs in European seabass domestication, we exploited published WGS data from three populations of wild European seabass [50, 51]. Using an identical pipeline to that applied with the farmed seabass SV dataset, we identified 38,408 high-confidence SVs in these samples (Additional file 14: Table 14). We will fully report this wild European seabass SV dataset in an independent study with distinct aim. Here, we focus on 41,336 SVs sharing identical locations in the reference genome derived from both datasets (see Methods) (Additional file 15: Table 15). For the current analysis, we compared genotypes for these merged SVs in samples comprising wild and farmed populations (Fig. 5).
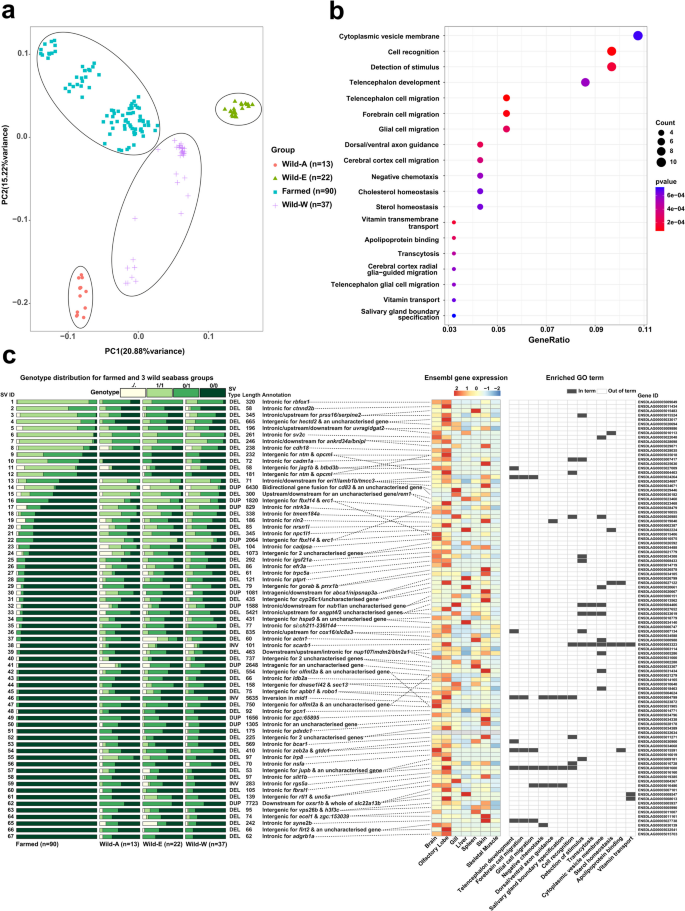
Comparative analysis of SVs in farmed and wild European seabass. These analyses were done using 41,336 SVs for n = 162 samples representing farmed and wild fish. The wild samples were from three geographical regions: Atlantic Ocean (Wild-A, n = 13), East Mediterranean (Wild-E, n = 22), West Mediterranean (Wild-W, n = 37). a PCA analysis of all SVs. b Enriched GO terms (q value/adjusted p value < 0.1) for 99 genes associated with 67 genetically differentiated SVs (top 5% FST values) common to the three comparisons: Farmed (n = 90) vs. Wild-A, Wild-E, or Wild-W. c Annotation of the 67 SVs and 99 associated genes. Left: relative SV genotype counts for each farmed vs. wild comparison along with annotations regarding SV type, length, genomic location and association to genes. Right: gene transcript per million (TPM) expression across a panel of tissues (standardized/scaled expression from −2 to + 2), and membership to enriched GO terms. Supporting results provided in Additional files Tables 14–18
Initially, we performed a principal component analysis (PCA) on the full set of SV genotypes to test the ability of SVs to capture expected genetic relationships across the sampled populations and individuals (Fig. 5a). This revealed genetically distinct groups of wild European seabass from different geographical regions, separated along PC1 and PC2, namely: Atlantic Ocean (‘wild-A’), West Mediterranean (‘wild-W’), and East Mediterranean (‘wild-E’), shown elsewhere to form the same genetically distinct groups using SNPs [52]. The farmed population forms a distinct group in the PCA, with sub-groups separated along PC1 and 2 (Fig. 5a), consistent with the mixed origins of the study population [33]. Overall, this analysis provides confidence in the quality of our SV discovery and genotyping pipeline, with respect to its ability to recover known population structure observed elsewhere using SNPs.
We next calculated the fixation index (FST) using genotypes for the 41,336 SVs in three comparisons: i) farmed (n = 90) vs. Wild-A (n = 13), ii) farmed (n = 90) vs. Wild-E (n = 22) and iii) farmed (n = 90) vs. Wild-W (n = 37). We retained only SVs identified within the top 5% FST values in all three comparisons, which were filtered to retain 67 SVs showing the same direction of allele frequency change in each case (Additional file 16: Table 16). This strategy reduces the confounding effects of population structure between the wild geographical groups, which would have occurred in a global analysis of all farmed vs. all wild fish.
SnpEff annotation revealed that the 67 SVs were associated with 99 unique genes (Additional file 16: Table 16). We performed a GO analysis using these genes (Fig. 5b; Additional file 17: Table 17), mapped differences in genotype frequency for the associated SVs in the three farmed vs. wild comparisons, quantified each gene’s tissue-specific expression (Fig. 5c) and summarised the known roles of orthologs from work in other species (Additional file 18: Table 18).
GO analysis revealed enrichment of the terms “Telencephalon cell migration”, “Telencephalon development”, “Cerebral cortex cell migration” and “Telencephalon glial cell migration” (Fig. 5b; Additional file 17: Table 17). The telencephalon (i.e., cerebrum/forebrain, which includes the cerebral cortex) is centrally involved in behaviour, neural plasticity and cognition in fish [53] and a primary target for domestication in diverse farmed animal species [54, 55]. Telencephalon-associated enriched GO terms were explained by an overlapping set of eight genes, including slit1b, encoding slit Homolog 1, involved in axon guidance and neural development, playing a key role in directing neuron migration and connectivity [56], as well as stress and behavioural responses [57]. A 97 bp intronic deletion in slit1b was present in moderate allele frequencies in each wild population (0.15, 0.19, 0.13 for Wild-A, Wild-E, Wild-W, respectively), but absent in the farmed population. robo1 was another gene explaining enrichment of telencephalon-associated GO terms (Fig. 5c), encoding roundabout guidance receptor 1, which mediates nerve axon guidance through its interaction with Slit proteins, and is crucial for neural circuit formation and development [58]. However, it should be noted that the 75 bp deletion associated with robo1 was ~ 77 kb upstream (Additional file 16: Table 16). Similar to slit1b, this SV was almost absent in the farmed population, but present in moderate frequencies in the wild groups (0.25, 0.30, 0.15 for Wild-A, Wild-E, Wild-W, respectively). It is notable that vertebrate Slit proteins form complexes with Robo proteins to dictate neuron axon guidance in the olfactory bulb of the telencephalon [59], as slit1b and robo1 were specifically expressed in the seabass olfactory lobe (Fig. 5c). Another gene explaining the telencephalon-associated enriched GO terms was lrp8, encoding low-density Lipoprotein receptor-related protein 8, which was also highly expressed in the seabass brain and olfactory lobe (Fig. 5c). LRP8 is the receptor for Reelin, an interaction that activates signalling pathways essential to synaptic plasticity, learning and memory formation [60]. A 97 bp intronic deletion within lrp8 was nearly absent in farmed fish, but present at moderate to high frequencies in the wild groups (0.63, 0.41, 0.15 for Wild-A, Wild-E, Wild-W, respectively). Interestingly, an intronic deletion in lrp8 showed significantly different allele frequencies between farmed and wild Atlantic salmon [19], hinting at convergent evolution.
Beyond genes explaining enrichment of the telencephalon-associated GO terms, many others within the 99 domestication-associated genes have roles associated with the brain and nervous system, including synapses and behaviour (Fig. 5c; Additional file 18: Table 18). For example, cadm1a, contributing to enriched term “detection of stimulus”, was expressed highly in the brain and olfactory lobe (Fig. 5c), consistent with its role as a synapse adhesion molecule [61]. A 78 bp intronic deletion in cadm1 showed an increase in allele frequency in the farmed seabass population (0.58) compared to the wild groups (0.08, 0.11 and 0.25 for Wild-A, Wild-E, Wild-W, respectively). While not contributing to any enriched terms, a 320 intronic deletion in rbfox1, encoding RNA splicing factor RNA Binding Fox-1 Homolog 1, was highly differentiated between farmed and wild populations, being almost fixed in the former (allele frequency: 0.95) (Fig. 5c). Again, rbfox1 was specifically expressed in the brain and olfactory lobe (Fig. 5c), consistent with the human ortholog’s known role as a neuron-specific factor linked to a range of neurological conditions, including autism [62]. Further genes associated with SVs showing genetic differentiation between wild and farmed seabass and expressed specifically in brain and/or olfactory lobe included: ctnnd2 (harboring a 58 bp intronic deletion), encoding adhesive junction associated synaptic protein—linked to neurodevelopmental disorders in humans, which regulates forebrain neuronal organisation and behavioural phenotypes in zebrafish [63]; sv2 (harboring a 261 bp intronic deletion) encoding a synaptic vesicle protein; btbd3b, which in mammals controls the orientation of neuron dendrites in the cerebral cortex [64]; nrsn1l, a neuron-specific gene encoding a protein supporting axon and dendrite extension [65]; and nsf, encoding a protein with multifaceted synaptic and neuronal functions [66]; among other examples (Fig. 5c; Additional file 18: Table 18).
Beyond the signal of brain-associated functions among the 99 genes, a small number of additional enriched terms are interesting in the context of domestication. Contributing to enriched term ‘cholesterol homeostasis’, we identified a 345 bp intronic deletion in npc1l1, present in much higher frequencies in the wild groups (Fig. 5c). In humans, this gene plays a key role in regulating hepatic cholesterol [67], consistent with its liver-specific expression in seabass (Fig. 5c). Several other genes impacted by the 67 SVs had roles in lipid storage, transport or metabolism, including abca1 and dgat2 (Additional file 18: Table 18).
Two of the 67 SVs were annotated as high-impact variants by SnpEff, while only one overlapped an evolutionary conserved GERP region (Additional file 16: Table 16). One of the high-impact SVs was a 6,430 bp duplication that overlapped two genes, spanning a non-coding exon and intronic region (containing a predicted enhancer) of cd83, as well as the last coding exon of a gene that is uncharacterized in Ensembl (ENSDLAG00005030182), but named as tripartite motif-containing protein 16-like by NCBI. This duplication is predicted to cause a bidirectional gene fusion. Both genes are most highly expressed in the seabass gill and the duplication is present at a much lower frequency in the farmed population (Fig. 5c). In other teleost species, cd83 has an immunological function in antigen-presenting dendritic cells in gills, and is strongly upregulated during vaccination and pathogen challenge [68, 69]. It will be interesting to test whether the identified duplication impacts immunological functions in European seabass carrying different genotypes.
Finally, we asked if any of the genetically differentiated SVs between wild and farmed seabass overlapped with Ensembl predicted regulatory elements. Among the 67 SVs, 6 overlapped with unique predicted regulatory elements, specifically 3 enhancers and 3 open chromatin regions. One open chromatin region (ENSDLAR00000234746) that shows predicted activity specifically during gastrulation, was disrupted by a 66 bp deletion located on CAJNNU010000003.1, present exclusively in wild seabass populations (allele frequency: 0.27, 0.19 and 0.10 for Wild-A, Wild-E, Wild-W, respectively). This region sits 90 Kb upstream of flrt2 (ENSDLAG00005030139), a conserved gene with key functions during early embryonic development, including heart morphogenesis [70]. Another notable SV was a 410 bp deletion on CAJNNU010000006.1, which disrupted an intronic enhancer (ENSDLAR00000182270) embedded in two overlapping genes, zeb2a (ENSDLAG00005030900) and gtdc1 (ENSDLAG00005034668), both with known roles in brain development and neurological functions [71, 72]. This SV was common in wild seabass (allele frequency: 0.33, 0.34 and 0.40 for Wild-A, Wild-E, Wild-W, respectively), but rare in the farmed population (allele frequency: 0.001). The remaining SVs were duplications, with one (6,430 bp deletion) discussed in the last paragraph (impacting cd83). We further identified a 1,656 bp duplication on CAJNNU010000016 that fully overlapped an open chromatin region (ENSDLAR00000252590) showing activity across embryonic development, found in the first exon of a poorly-characterised gene annotated as zgc:65,895 (ENSDLAG00005034796). This SV was again rare in the farmed population (allele frequency: 0.01) but common in each wild population (allele frequency: 0.27, 0.23 and 0.18 for Wild-A, Wild-E, Wild-W, respectively). Finally, a 1,588 bp duplication on scaffold CAJNNU010000021.1 fully overlapped with enhancer ENSDLAR00000140351, which shows predicted activity in gill, gonad, and liver. This enhancer is located in the first intron of nub1 (ENSDLAG00005006151), a gene encoding NEDD8 ultimate buster 1, an interferon stimulated molecule that downregulates NEDD8 [73], a gene encoding a ubiquitin-protein considered responsible for variation in resistance to infectious pancreatic necrosis in farmed Atlantic salmon [74]. Again, this duplication was rare in the farmed population (allele frequency: 0.02), but common in the wild populations (allele frequency: 0.21, 0.32 and 0.23 for Wild-A, Wild-E, Wild-W, respectively).
Collectively, these findings highlight potential functional impacts of SVs on key regulatory elements and associated genes, underscoring their relevance in understanding phenotypic divergence between wild and farmed populations.