Chemistry
The compounds were synthesized following the reported procedures [16]. (Z)−4-arylidene-2-substituted oxazol-5(4 H)-one intermediates (3) were prepared by Erlenmeyer method through reaction of N-acetyl glycine (1) or hippuric acid with the appropriate aldehyde (2) in acetic anhydride and sodium acetate. Kim Compounds (5a-d) were prepared by cyclocondensation reaction of (Z)−4-arylidene-2-substituted oxazol-5(4 H)-one intermediates (3) with 4-amino-N-benzylphenyl acetamide (4) in dry pyridine to yield the target molecules (5a-d) [16]. These four compounds are Z type and include 5a (KIM-161): (Z)-N-Benzyl-2-(4-(4-(4-methoxybenzylidene)−2-methyl-5-oxo-4,5-dihydro-1 H-imidazol-1-yl)phenyl)acetamide, 5b (KIM-111): (Z)-N-Benzyl-2-(4-(4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1 H-imidazol-1-yl)phenyl)acetamide, 5c(KIM-261): (Z)-N-Benzyl-2-(4-(4-(4-methoxybenzylidene)−5-oxo-2-phenyl-4,5-dihydro-1 H-imidazol-1-yl)phenyl)acetamide, 5 d (KIM-231): (Z)-N-Benzyl-2-(4-(4-(4-hydroxy-3-methoxybenzylidene)−5-oxo-2-phenyl-4,5-dihydro-1 H-imidazol-1-yl)phenyl)acetamide.
All the target compounds were confirmed by different analytical techniques including spectral analysis using nuclear magnetic resonance (NMR), and the molecular formulae were detected using high-resolution mass spectrometry (HRMS). The purity of all compounds was confirmed by liquid chromatography/mass spectrometry (LC/MS) to be higher than 95%. Scheme 1 shows the synthetic method of our target compounds (5a-d) and their project codes. The synthetic procedures and detailed characterization of all compounds have been comprehensively described in our earlier work [16].
The N-acetylglycine (1, 10 mmol), appropriate aldehyde (2, 10 mmol), acetic anhydride (1.9 ml, 2 equiv.) and sodium acetate (0.08 g, 0.1 equiv.) were heated at 80 °C for 30 min then cooled. The appropriate oxazolone (3, 2 mmol) was mixed with the amine (2-(4-aminophenyl)-N-benzylacetamide) (0.48 g, 2 mmol) in dry pyridine (6 ml) under inert atmosphere and was heated to 100 °C for 8 h to produce target compounds (5a-d)
Physicochemical properties
The physicochemical properties explained the molecular weights, molecular formula, number of heavy atoms, number of aromatic heavy atoms, fraction Csp3, number of rotatable bonds, number of hydrogen bonds, molar refractivity, topological polar surface area, and other physicochemical features shown in Table 1. They also displayed the high lipophilic characters of these compounds which are responsible for high GIT absorption and BBB permeation especially in the two compounds Kim-161, Kim-111. They also have moderate water solubility, good bioavailability score, moderate synthetic accessibility, and obey Lipiniski rule which means these compounds are suitable for production as drug. On the other side, the two compounds Kim-231, Kim-261 have poor water solubility, good bioavailability score, moderate synthetic accessibility, and have two violations from Lipiniski rule which means these compounds have no optimum characters for production as drug. Table 1 shows the physicochemical properties of the target compounds calculated by using SwissADME [29].
TPSA = topological polar surface area. Bioavailability image: The colored zone is the suitable physiochemical space for oral bioavailability: Lipo = lipophilicity, polar = polarity, insole = insolubility, insatu = instauration, Flex = flexibility.
Biological screening
MTT assay
A comparative investigation of proliferation inhibition across cancer cell line T24 by the MTT assay [30] for Kim-111, Kim-161, Kim-231, and Kim-261 molecules revealed that both Kim-111 and Kim-161 drugs exhibited cytotoxicity to the cells, demonstrating varying inhibition percentages with IC50 = 67.29 µM for Kim-111, and IC50 = 56.11 µM for Kim-161. The inhibitory impact considerably increased along with the increased concentrations in contrast to Kim-231, and Kim-261which exhibited no inhibitory effect at the concentrations tested in the current set up as illustrated in Fig. 3.
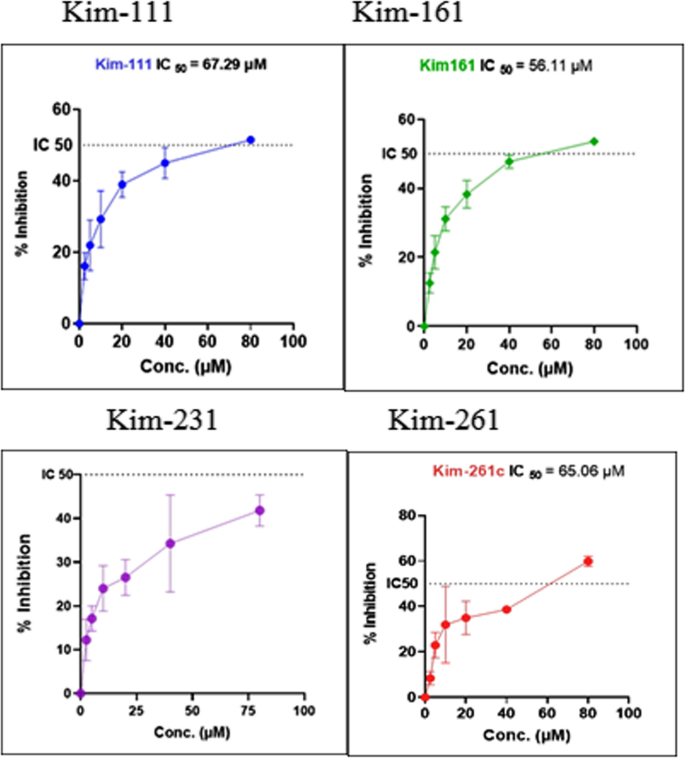
Anti-proliferation effect of Kim-111 (blue curve), Kim-161 (green curve), Kim-231 (violet curve), and Kim-261 (red curve) on the urinary bladder cancer cell line (T24)
Effect of Kim 111 and Kim 161 compounds on cell senescence and apoptosis in T24 urinary bladder cancer cells
A panel of suggested anticancer pathways were tested for evaluating the impact of Kim 111 and Kim 161 treatment on the genes expression responsible for cell senescence, apoptosis, inflammation and metastasis (Table 2).
The markers of responsible for cell senescence; (p53), oncogenesis (Kras) and cell apoptosis; (BAX and caspase 3) were evaluated to detect the difference between the Kim-treated groups and the control non-treated T24 bladder cancer cells.
P53 gene responsible for cell senescence, Kim 111 treatment showed statistically lower expression in comparison to the untreated T24 cells, but in contrast, the cells treated with Kim 161 showed a dramatic higher expression in comparison to the untreated cells. On a same pattern, treatment with Kim 111 reduced the expression of Kras oncogen significantly but Kim 161 showed statistically higher expression in comparison to the untreated cells. To evaluate the effect of the tested compounds on cell apoptosis, the genes suggested to regulate the apoptosis process were investigated following the T24 cells treatment, namely BAX and caspase 3. The data demonstrated that cells treated with Kim 111 and 161 demonstrated a higher expression with statistically significant difference between both treated groups, and in comparison, to the untreated cells (Fig. 4).
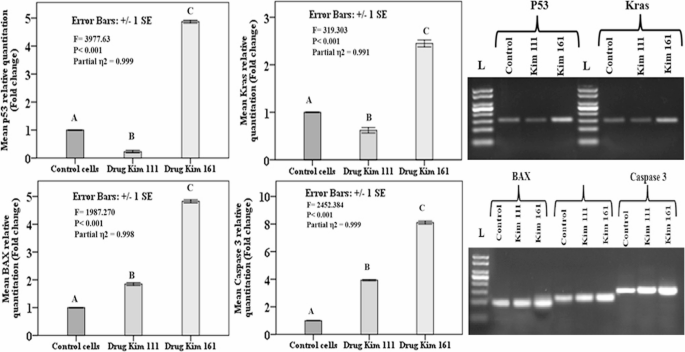
Relative quantitation (fold change) of the genes in T24 bladder cancer cell line from the three groups as studied by real-time PCR and gel electrophoresis. The figure shows the effect of IC50 of Kim 111 and Kim 161 compounds in comparison to the untreated cells on the genes expression responsible for cell senescence; (p53), oncogenes (Kras) and cell apoptosis; (BAX and caspase 3) (p53 qPCR product (151 bp) and Kras qPCR product (147 bp), BAX qPCR product (100 bp), and caspase 3 qPCR product (146 bp). Data presented as mean ± standard error of gene expression fold changes of cells′ triplicates. Capital letters indicate p values ≤ 0.05 using One-Way ANOVA with Tukey post-hoc testing (similar letters indicate non-significant difference, different letters denote significant difference)
Effect of Kim 111 and Kim 161 compounds on the inflammatory markers in T24 urinary bladder cancer cells
Regarding the inflammatory genes, IL6 showed statistically lower expression in the cells treated with Kim 111 in comparison to the untreated cells, but in contrast, the cells treated with Kim 161 showed statistically higher expression in comparison to the untreated cells. TNFα and NF-κB1 genes expression increased following the treatment with Kim 111 and Kim116, with statistically significant difference between both treated groups, and in comparison, to the untreated cells (Fig. 5).
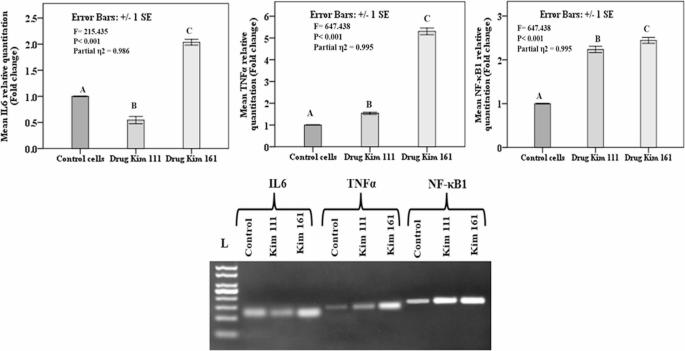
Relative quantitation (fold change) of the inflammatory genes Interleukin 6 (IL6), Tumor Necrosis Factor alpha (TNFα), and Nuclear Factor kappa B (NF-κB) in T24 bladder cancer cell line by real-time PCR and gel electrophoresis. (IL6 qPCR product (132 bp), TNFα qPCR product (135 bp), and NF-κB1 qPCR product (161 bp)). Data presented as mean ± standard error of gene expression fold changes of cells′ triplicates. Capital letters indicate p values ≤ 0.05 using One-Way ANOVA with Tukey post-hoc testing (similar letters indicate non-significant difference, different letters denote significant difference)
Effect of Kim 111 and Kim 161 compounds on the autophagy and metastasis markers in T24 urinary bladder cancer cells
The current study assessed the role of Kim 111 and Kim 161 in regulating cell proliferation and survival through evaluating the phosphoinositide 3-kinase (PIK3CA) and its target genes; Akt, and mTOR. Both the Kim 111 and Kim 161- treated groups showed significantly lower PIK3CA and mTOR expression relative to the untreated group. There was no statistically-significant difference in the expression of the PIK3CA and mTOR genes between the cells treated with Kim 111 and those treated with Kim 161. In contrast, Akt gene expression showed higher expression with statistically significant difference between both treated groups, and in comparison, to the untreated cells. Regarding the metastatic gene Matrix metalloproteinase-9 (MMP-9), both treated groups showed statistically lower expression in comparison to the untreated cells, with a non-significant difference between cells treated with Kim 111 and Kim 161 (Fig. 6).
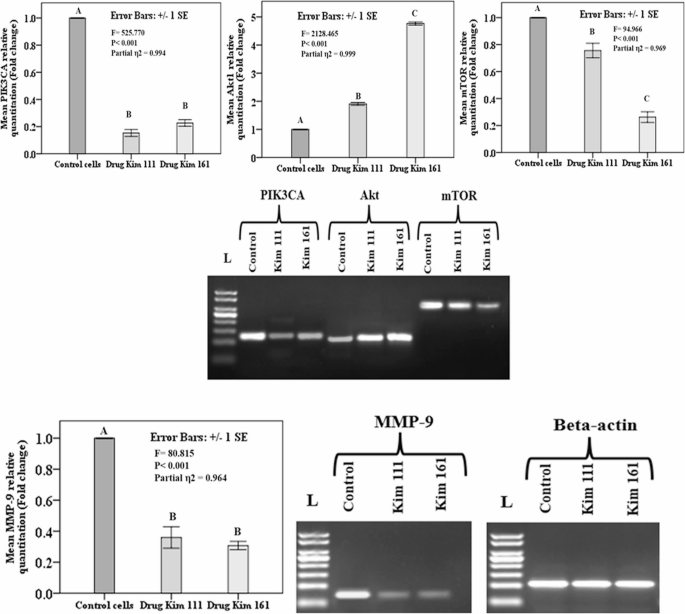
Relative quantitation (fold change) of the genes regulating cell proliferation, survival and motility (phosphoinositide 3-kinase signaling (PIK3CA) and its targets protein kinase Akt and mammalian target of rapamycin (mTOR) and Matrix metalloproteinase-9 (MMP-9) in T24 bladder cancer cell line by real-time PCR and gel electrophoresis. PIK3CA qPCR product (128 bp), Akt qPCR product (113 bp), and mTOR qPCR product (318 bp), MMP-9 qPCR product (79 bp). β-actin qPCR product (104 bp). Β-actin was used as a housekeeping gene. L= 50 bp ladder. Data presented as mean ± standard error of gene expression fold changes of cells′ triplicates. Capital letters indicate p values ≤ 0.05 using One-Way ANOVA with Tukey post-hoc testing (similar letters indicate non-significant difference, different letters denote significant difference)
Data presented as mean ± standard error of gene expression fold changes of cells′ triplicates. Capital letters indicate p values ≤ 0.05 using One-Way ANOVA with Tukey post-hoc testing (similar letters indicate non-significant difference, different letters denote significant difference).
The above-mentioned data demonstrated the potential of novel imidazole derivatives, Kim-161 and Kim-111, as promising anticancer agents against urothelial carcinoma (T24 cells). Clinicians may consider Kim-161 over Venetoclax if ongoing trials confirm enhanced BCL-2 inhibition, improved safety, or efficacy in resistant cancers. We hope its optimized structure offers better pharmacokinetics, reduced toxicity, or broader antitumor activity, addressing Venetoclax limitations. However, further studies are needed to validate these potential advantages.
Computational modeling
Docking and binding poses
The top three targets identified (PTK6, FLT3, and BCL-2) correspond to the crystal structures 6CZ4, 4XUF, and 6O0K, respectively. The targets PTK6 (6CZ4), FLT3 (4XUF), and BCL-2 (6O0K) were selected due to their critical roles in urothelial carcinoma (UC) progression. PTK6 promotes cell proliferation and survival, making it a key oncogenic kinase. FLT3, primarily associated with leukemia, is implicated in UC through aberrant signaling pathways. BCL-2, an anti-apoptotic protein, is often overexpressed in UC, contributing to chemoresistance. The crystal structures 6CZ4, 4XUF, and 6O0K provide high-resolution templates for docking studies, enabling the evaluation of novel derivatives as potential inhibitors. Targeting these proteins may disrupt proliferation and survival mechanisms, offering a strategic approach for UC therapy. Glide XP docking scores for Kim-111 and Kim-161 in these sites are summarized in Table 3 alongside the scores of the native co-crystallized ligands. Overall, Kim-111 showed very favorable docking to the two kinases PTK6 and FLT3 (XP GScore ≈ − 11.3 and − 11.6 kcal/mol, respectively), essentially matching the binding score of the native inhibitors (–14.76 for PTK6’s ligand FKY, − 11.75 for FLT3’s Quizartinib). Kim-161 likewise docked well in those sites (≈ − 11.6 kcal/mol). In BCL-2, the docking scores for Kim-111 and Kim-161 were more modest (around − 7.4 to − 7.9 kcal/mol), reflecting weaker predicted binding than the native BCL-2 inhibitor Venetoclax (docking score − 10.94 kcal/mol). Notably, the poses of Kim-161 in BCL-2 overlapped substantially with Venetoclax’s position, indicating Kim-161 can fit in the same hydrophobic pocket that accommodates the BH3-domain of pro-apoptotic proteins. Figure 7 shows the 3D interaction diagram of Venetoclax bound in the BCL-2 (6O0K) pocket after 100 ns MD, however Fig. 8 shows the 3D interaction diagram of Kim-161 bound in the BCL-2 (6O0K) pocket after 100 ns MD. Key interacting residues are highlighted with their interaction frequency over the trajectory. Kim-161 forms strong hydrogen bonds (purple dashed arrows) with Tyr108 (83% occupancy) and Asn143 (66%), mimicking the interactions of the native inhibitor. Hydrophobic contacts (green lines) with Phe104 (57%) and Met115 stabilize the ligand in the hydrophobic groove. These interactions help anchor Kim-161 similarly to Venetoclax, supporting a BCL-2 inhibitory mechanism.
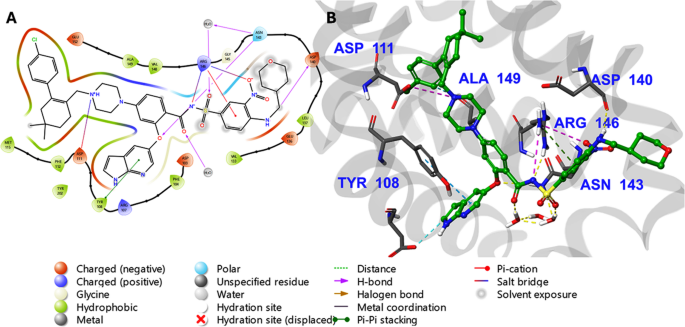
3D interaction diagram of Venetoclax bound in the BCL-2 (6O0K) pocket after 100 ns MD
The induced fit docking (IFD) results showed further optimization of these poses. Allowing side-chain flexibility led to small improvements in docking scores for the Kim compounds (Table 3). For instance, in FLT3 the IFD-refined pose of Kim-111 achieved a docking score of − 14.29 kcal/mol (improved from − 11.58), surpassing the native ligand’s re-dock score. Such improvements suggest Kim-111 can induce slight conformational adjustments in the FLT3 binding site to enhance complementarity. In BCL-2, IFD refinement of Kim-161 allowed Tyr108 and Arg146 side chains to shift and form the above-mentioned hydrogen bonds, yielding a more favorable score (–8.84 vs. initial − 7.89). Overall, IFD confirmed that Kim-111 and Kim-161 are capable of binding in the active sites of these targets with only minor local receptor adjustments, and no major backbone motion was required to accommodate them.
Molecular Mechanics Generalized Born Surface Area (MM-GBSA) was used to estimate the binding free energy because it is considered an ideal tool for high-throughput screening to determine structural stability, and predict binding affinities. Being a faster and less expensive computational tool makes it a promising alternative to the more complex free energy calculations and empirical scoring functions.
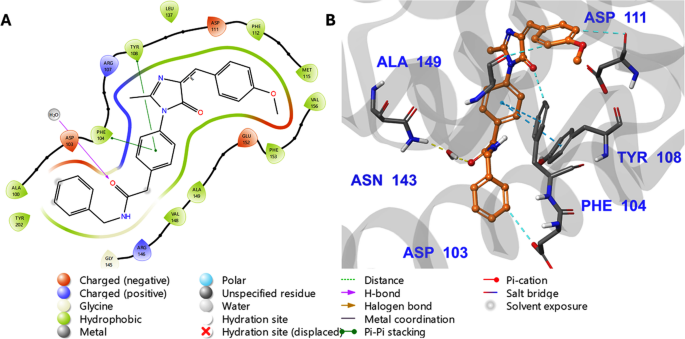
3D interaction diagram of Kim-161 bound in the BCL-2 (6O0K) pocket after 100 ns MD
Table 3 displays the docking scores (Glide XP) and (MM-GBSA) binding energies for Kim-161 and Kim-111 in top targets, compared to native ligands.
Docking scores (in kcal/mol) are from Glide XP (negative values indicate better predicted affinity). ΔGbind is the binding free energy from Prime MM-GBSA (more negative = more favorable). PTK6 native Ligand refers to PDB 6CZ4’s inhibitor (FKY); FLT3 native is Quizartinib in 4XUF; BCL-2 native is Venetoclax in 6O0K. Figure 9 shows the 3D interaction diagram of Kim-111 bound in the BCL-2 (6O0K) pocket after 100 ns MD.
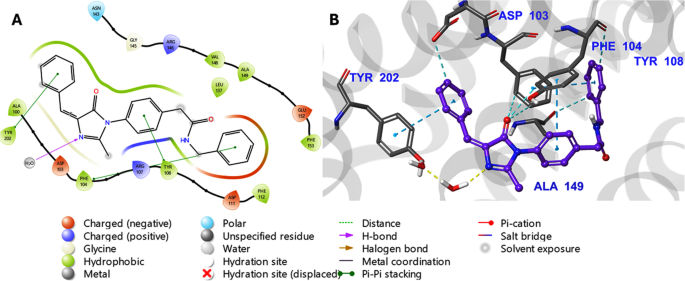
3D interaction diagram of Kim-111 bound in the BCL-2 (6O0K) pocket after 100 ns MD
As shown in Table 3, the MM-GBSA calculations followed the trend of the docking scores. Kim-111 and Kim-161 were predicted to bind most strongly to FLT3 and PTK6 (ΔGbind ~ − 69 to − 75 kcal/mol), whereas their binding to BCL-2 was relatively weaker (ΔGbind ~ − 66 kcal/mol). Importantly, all ΔGbind values are significantly negative, suggesting that both compounds can form stable complexes with each target in an aqueous environment. The native ligands, being high-affinity inhibitors, have substantially more favorable ΔGbind (e.g., − 122.5 kcal/mol for Venetoclax with BCL-2), which is expected given they were optimized for those targets. Figure 10 illustrates the comparative binding free energies for the two compounds vs. native ligands, highlighting that Kim-111 and Kim-161 achieve strong binding in the kinases and moderately strong binding in BCL-2.
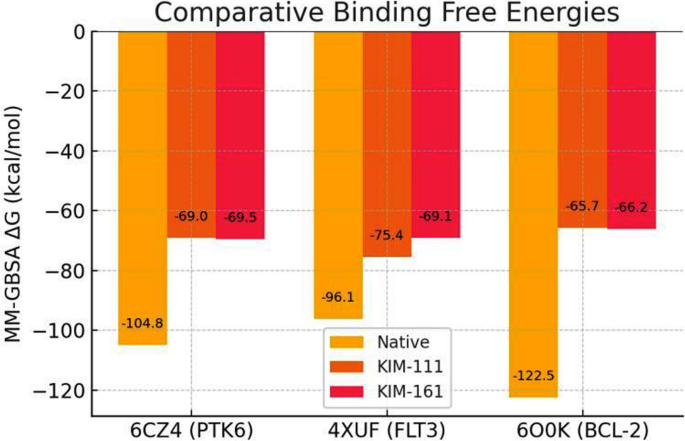
Comparative binding free energies (Prime MM-GBSA ΔGbind) for Kim-111 (orange) and Kim-161 (red) versus native co-crystallized ligands (yellow) in three target proteins. More negative bars indicate stronger binding affinity. Native inhibitors (yellow) show the most favorable energies in each target (e.g., –122.5 kcal/mol for Venetoclax in BCL-2, left cluster). Kim-111 and Kim-161 exhibit substantial binding to FLT3 (middle cluster) and PTK6 (left cluster) with ΔGbind around –70 kcal/mol, comparable to the native ligands. In BCL-2 (right cluster), Kim-111 and Kim-161 have less negative ΔGbind (~–66 kcal/mol) relative to Venetoclax but still indicate favorable binding. These results corroborate the docking scores, suggesting Kim-111 may bind FLT3 and PTK6 slightly more strongly than Kim-161, whereas both compounds bind BCL-2 with similar moderate affinity
Molecular dynamics stability
The 100 ns MD simulations provided dynamic validation of the docking poses. In all cases, the protein–ligand complexes remained intact over the simulation, with no unbinding events observed. The RMSD profiles (protein Cα and ligand) are shown in Fig. 11 for a representative complex, and quantitative stability metrics from all simulations are summarized in Table 3. Overall, the FLT3 complexes were the most stable: with Kim-111 bound to FLT3, the protein backbone RMSD plateaued around ~ 1.8 Å and the Ligand RMSD stayed below 1.5 Å for nearly the entire 100 ns (Fig. 11). Kim-161 in FLT3 showed a similarly low ligand RMSD (~ 1.0 Å average), indicating that both compounds snugly occupy the FLT3 kinase site without significant displacement. The PTK6 simulations showed a slightly higher backbone RMSD (~ 2.5 Å) but still reasonable stability. Notably, Kim-111 in PTK6 maintained a low ligand RMSD (~ 2 Å), whereas Kim-161 in PTK6 had larger fluctuations (ligand RMSD rising to ~ 4 Å transiently), suggesting Kim-161’s fit in PTK6 is less optimal or stable than Kim-111’s.
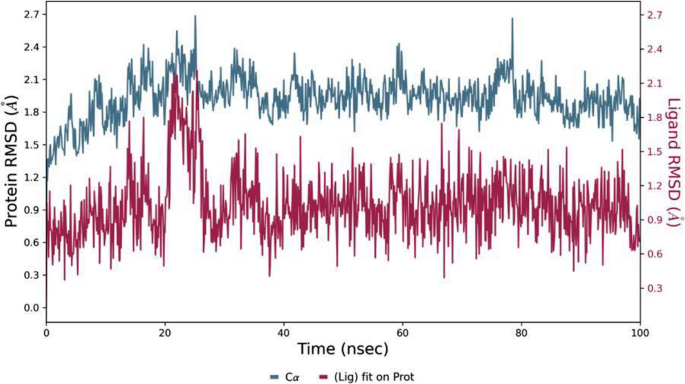
RMSD versus time for the FLT3–Kim-111 complex (PDB 4XUF) during a 100 ns MD simulation. The protein’s Cα RMSD (blue) stabilizes around 1.8–2.4 Å, indicating an equilibrated protein structure. The ligand Kim-111’s RMSD (magenta, calculated after fitting on the protein) remains low (~0.5–1.5 Å) throughout the simulation, reflecting that Kim-111 stays tightly bound in the FLT3 active site without significant drift. This high stability suggests a well-maintained protein–ligand interaction network
In the BCL-2 simulations, the behavior of the two compounds diverged. Kim-161 formed a very stable complex with BCL-2: after an initial adjustment (ligand RMSD < 2 Å in the first 5 ns), Kim-161 remained close to its docked position (average ligand RMSD ~ 1.8 Å) for the rest of the trajectory. In contrast, Kim-111 in BCL-2 exhibited more movement – its Ligand RMSD fluctuated between 2 and 4 Å, albeit remaining in the pocket. This aligns with the weaker docking score and MM-GBSA energy of Kim-111 for BCL-2, suggesting Kim-111 does not engage BCL-2 as firmly as Kim-161 does. The BCL-2 protein itself remained stable (backbone RMSD ~ 1.5 Å) when either ligand was bound, comparable to the control simulation with Venetoclax (which showed backbone RMSD ~ 1.3 Å and ligand RMSD ~ 1 Å, as expected for a very tight binder).
The MD analyses also examined residual flexibility and interactions. RMSF plots (see Supplementary Figures S1) showed that loop regions at the periphery of each binding site had mild fluctuations (up to 3 Å), but critical binding site residues remained relatively rigid (RMSF < 1 Å) in the presence of each ligand. In FLT3, residues in the activation loop and the hinge (which interact with inhibitors) were stabilized by both Kim-111 and Kim-161, like the native inhibitor. In BCL-2, the presence of Kim-161 kept the binding groove residues (e.g., Tyr108, Phe104, Arg146) ordered, whereas with Kim-111 there was slightly more fluctuation in the loop containing Arg146, correlating with Kim-111’s less consistent interactions there.
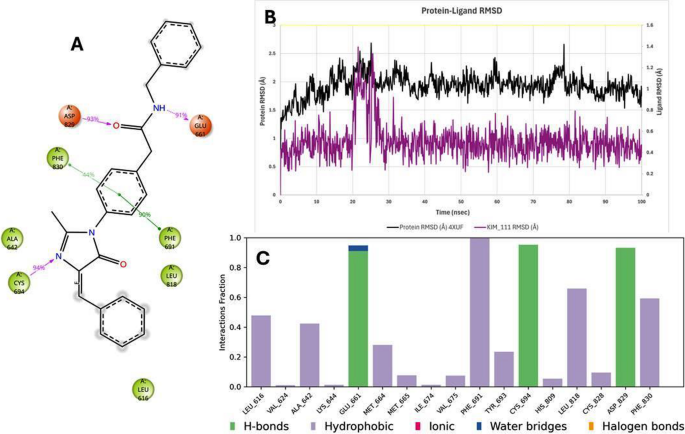
Crucially, the MD trajectories confirmed that key protein–ligand interactions identified in docking persisted over time. For example, in the BCL-2–Kim-161 simulation, the hydrogen bond between Kim-161’s amide carbonyl and the side chain of Asn143 was maintained ~ 66% of the time, and a stable hydrogen bond (83% occupancy) formed between Kim-161’s secondary amine and the backbone carbonyl of Tyr108. These interactions closely mirror those formed by Venetoclax in BCL-2, lending confidence that Kim-161 indeed engages the BCL-2 pocket in a functionally relevant manner. Kim-111 also retained a hydrogen bond with Tyr108 but with lower frequency (~ 40% of frames), explaining its weaker binding. In FLT3, Kim-111 consistently formed two hydrogen bonds with the kinase hinge region (to a backbone carbonyl and a side-chain of a hinge residue, analogous to Quizartinib’s interactions), which remained > 70% occupied during MD. Kim-161 in FLT3 also maintained hinge region hydrogen bonds, though one interaction was intermittent (about 30% occupancy), consistent with its slightly lower affinity prediction. Hydrophobic contacts were largely preserved, e.g., Kim-111 stayed nestled against FLT3’s gatekeeper residue and Phe830 (in the activation loop) through π-stacking and van der Waals contacts, and Kim-161 remained in contact with BCL-2’s hydrophobic groove residues Phe104 and Val130 over > 80% of the simulation frames. Figures 12 and 13 display the 2D interaction diagram of Kim-161, Kim-111 bound in the FLT-3 (4XUF) pocket after 100 ns MD and molecular dynamic analysis graphs.
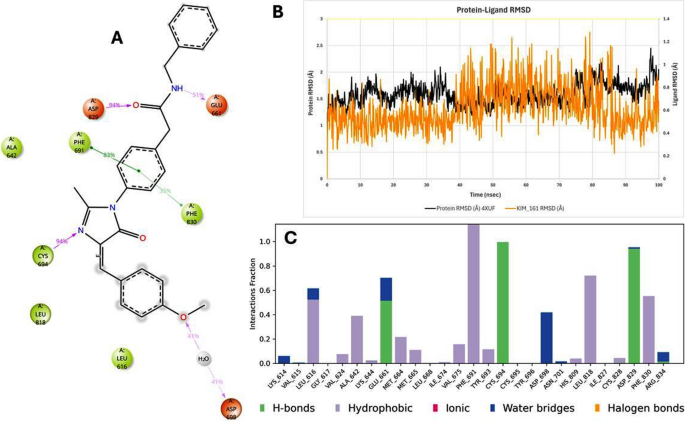
2D interaction diagram of Kim-161 bound in the FLT-3 (4XUF) pocket after 100 ns MD and molecular dynamic analysis graphs
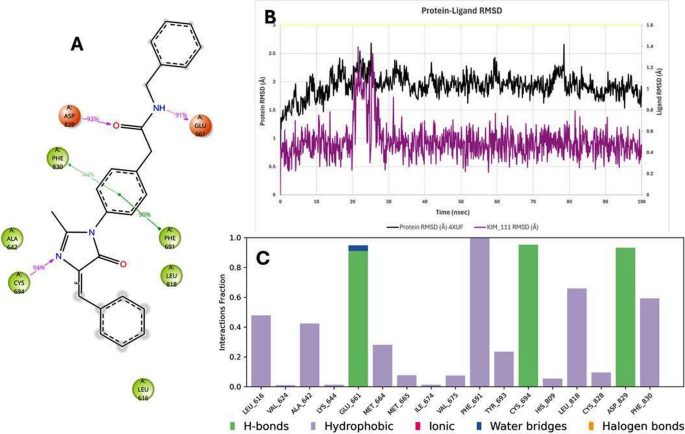
2D interaction diagram of Kim-111 bound in the FLT-3 (4XUF) pocket after 100 ns MD and molecular dynamic analysis graphs
In summary, the MD results support the docking findings by demonstrating that Kim-111 and Kim-161 form stable complexes with all three putative targets in an explicit solvent environment. The stability ranking from MD (most stable in FLT3, followed by PTK6, then BCL-2 for Kim-111; and FLT3 ≈ BCL-2 > PTK6 for Kim-161) aligns with the binding affinity predictions. Minor differences in stability (e.g., Kim-161 outperforming Kim-111 in BCL-2, and Kim-111 slightly more stable in PTK6) were observed, highlighting the complementary nature of these two compounds in targeting different proteins. The persistence of critical hydrogen bonds and the low ligand RMSDs especially underscore that Kim-111 is well-suited for targeting FLT3/PTK6, whereas Kim-161 is particularly effective at engaging BCL-2.
Computational modeling provides a mechanistic rationale for the biological activities of Kim-111 and Kim-161. Docking and MD simulations identified multiple potential targets for these compounds, which suggests a polypharmacological mode of action (i.e. hitting more than one target) [31]. Two oncogenic kinases (PTK6 and FLT3) and the anti-apoptotic protein BCL-2 emerged as top binding targets. This aligns well with the experimental observations that Kim-111 and Kim-161 exhibit potent anti-cancer effects in bladder cancer cells, likely through a combination of pro-apoptotic and anti-proliferative mechanisms.
BCL-2 as an apoptosis-related target: The strong binding of Kim-161 to BCL-2 is especially notable. BCL-2 is a key regulator of cell death; inhibition of BCL-2 frees pro-apoptotic factors like BAX/BAK to trigger mitochondrial apoptosis. Our simulations showed that Kim-161 occupies the same site as the known BCL-2 inhibitor Venetoclax, even forming analogous interactions (Tyr108 and Asn143 hydrogen bonds, hydrophobic contact with Phe104). Although Kim-161’s computed affinity is lower than Venetoclax, it is still substantial, and the MD data confirmed the complex remains stable. This provides a molecular basis for the apoptosis induction observed in treated cells – if Kim-161 binds BCL-2, it would neutralize BCL-2’s anti-apoptotic function, leading to increased apoptosis. Indeed, in our biological assays, both compounds increased the expression of pro-apoptotic genes (BAX, Caspase 3) and markers of cell death in bladder cancer cells, consistent with BCL-2 inhibition relieving apoptosis suppression. Interestingly, Kim-161 had a greater effect on these apoptosis markers than Kim-111 (e.g., higher BAX, caspase-3, and p53 expression in Kim-161–treated cells, relative to Kim-111), which correlates with Kim-161’s stronger BCL-2 engagement in silico. We therefore propose that Kim-161 acts as a BH3-mimetic agent targeting BCL-2, triggering intrinsic apoptotic pathways in the cancer cells.
Kinase inhibition and anti-proliferative effects: Both Kim-111 and Kim-161 showed high docking affinity to PTK6 (protein tyrosine kinase 6) and FLT3 (Fms-like tyrosine kinase 3). These kinases are implicated in tumor cell proliferation and survival signaling [30,31,32]. PTK6, for instance, is overexpressed in some cancers and can promote migration and growth, while FLT3 activates pathways like PI3K/Akt and Ras/MAPK in leukemia and potentially other contexts [33,34,35]. Our finding that Kim-111 binds very stably to FLT3 (with low RMSD and persistent hydrogen bonds in the ATP-binding site) suggests Kim-111 could be an effective FLT3 inhibitor. Kim-161 also bound FLT3 well, albeit slightly less tightly. In the biological data, we observed downregulation of PI3K (PIK3CA gene) and mTOR expression in cells treated with either compound, along with changes in Akt and KRAS expression. These could be downstream consequences of kinase inhibition: if Kim-111 and Kim-161 inhibit FLT3 or PTK6, they would dampen PI3K/Akt and RAS signaling, resulting in reduced proliferation and possibly induction of senescence. Notably, Kim-111-treated cells had lower p53 and KRAS expression than controls (indicating a senescence-like cell cycle arrest), whereas Kim-161-treated cells showed an opposite trend for those genes. This divergence might be explained by their target spectrum Kim-111, with stronger kinase (FLT3/PTK6) inhibition, may enforce cell-cycle arrest without heavily stressing apoptotic pathways (hence lower p53 in a feedback loop), while Kim-161, by strongly hitting BCL-2, pushes cells into apoptosis (which can elevate p53 as a DNA damage response). In essence, Kim-111 might act more as a multi-kinase inhibitor, slowing proliferation, whereas Kim-161 has a dual action: moderate kinase inhibition plus potent apoptosis induction via BCL-2. Such multi-targeted kinase inhibitors are common in oncology (e.g., many TKIs hit multiple kinases) [16], and can be advantageous by blocking redundant survival pathways.
Target selectivity and synergy: The differential target binding also provides insight into how Kim-111 and Kim-161 could be used therapeutically. Kim-111’s high affinity for FLT3 suggests potential in malignancies where FLT3 is important (e.g. leukemia), or in solid tumors if FLT3 or PTK6 contribute to growth. Meanwhile, Kim-161’s targeting of BCL-2 indicates it could sensitize cancer cells to apoptosis or even be combined with other therapies. In bladder cancer, BCL-2 is one mechanism of chemo-resistance; thus Kim-161 might enhance the efficacy of chemotherapy by disabling tumor cell survival programs. The fact that both compounds can engage multiple targets (kinase and BCL-2) is promising, as cancer cells often rely on network redundancy. By concurrently attacking survival signaling and anti-apoptotic defenses, Kim-111 and Kim-161 may achieve a one-two punch: forcing cell cycle exit and then driving programmed cell death. This hypothesis is supported by the comprehensive changes seen in gene expression (senescence markers, apoptotic markers, and inflammatory pathways all altered). Our computational data back up this breadth of action with concrete binding interactions at the molecular level. Figures 14, and 15 show the 2D interaction diagram of Kim-161 and Kim-111 bound in the FLT-3 (4XUF) pocket after 100 ns MD and molecular dynamic analysis graphs.
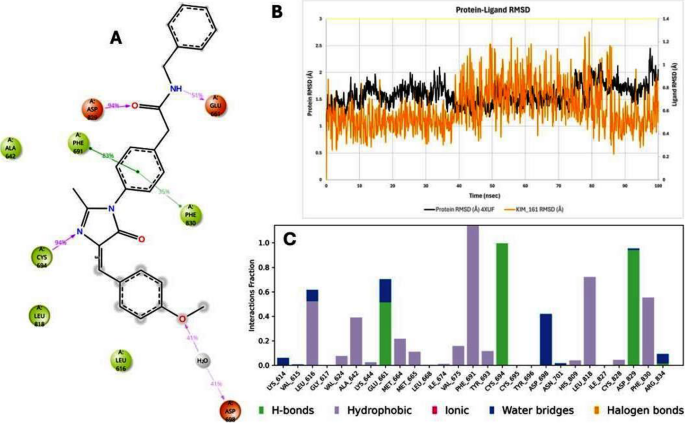
2D interaction diagram of Kim-161 bound in the FLT-3 (4XUF) pocket after 100 ns MD and molecular dynamic analysis graphs
Given that both Kim-111 and Kim-161 are designed as substituted imidazole derivatives with activity against numerous kinases, it is crucial to consider their potential off-target effects, which may arise from unintended interactions with non-target kinases or cellular proteins. These effects raise concern about the therapeutic efficacy, safety and specificity of these compounds [36].
2D interaction diagram of Kim-161 bound in the FLT-3 (4XUF) pocket after 100 ns MD and molecular dynamic analysis graphs
Since the kinase inhibitors often share conserved ATP-binding sites, cross reactivity and or/interactions with non-target kinases. For example, KIM-161 has been reported to downregulated several other kinases such as the ERK1/2, GSK-3α/β, HSP27, and JAK/STAT2 signals which may result in unintended immune modulation [16].
Moreover, the imidazole derivatives are known to interact with cytochrome P450 enzymes, potentially affecting the drug pharmacokinetic and metabolism. The hydrogen binding potential of those derivatives is another factor that raises the possibility of binding to ion channels or G-protein coupled receptors (GPCRs), which could manifest as neurological, endocrinal or gastrointestinal disturbances [37].
Thus, future studies should incorporate in-vivo toxicity assays, selectivity tests and wide molecular docking profiling for a broad panel of kinases to predict any systemic risks and mitigate any side effects to ensure the therapeutic efficacy and safety of those compounds.
The in silico findings from this study should also be followed by targeted experimental validation. For instance, pull-down or SPR assays could confirm direct binding of Kim-161 to BCL-2, and kinase inhibition assays (or cellular phospho-substrate readouts) could verify if FLT3 or PTK6 activity is suppressed by Kim-111. Co-crystallization or cryo-EM of Kim-161 with BCL-2 would definitively show if it bound in the Venetoclax pocket as predicted. Likewise, co-crystal structures with FLT3 or PTK6 would guide optimization of the scaffold for even tighter binding as our modeling suggests, for example, that adding functionality to interact with PTK6’s Asp164 (hinge region) might boost Kim-161’s affinity for PTK6. Medicinal chemistry optimization could proceed differently for the two compounds: for Kim-161, increasing affinity to BCL-2 (perhaps by extending into the P2 pocket like Venetoclax’s chlorophenyl group) could yield a potent apoptotic agent, whereas for Kim-111, maintaining broad kinase coverage but improving BCL-2 activity might create a balanced dual-target drug. Given the computational insight that each compound excels on different targets, designing analogs that combine the favorable features of both (kinase inhibition of Kim-111 with the BCL-2 inhibition of Kim-161) is an intriguing strategy.
In conclusion, our computational modeling study provides a detailed picture of how Kim-111 and Kim-161 likely interact with cancer-related proteins at the molecular level. The data supports a mechanism where Kim-161 directly antagonizes BCL-2, unleashing apoptosis, while Kim-111 potently inhibits oncogenic kinases like FLT3/PTK6, impeding survival signaling and both compounds exhibit overlapping activities across these targets. This dual modality is highly consistent with the observed anti-proliferative and pro-apoptotic effects in bladder cancer cells. The most promising target identified is BCL-2, given its pivotal role in apoptosis and the strong evidence of Kim-161 binding; targeting BCL-2 aligns with the pronounced cell death (and upregulation of apoptosis markers) seen experimentally. PTK6 and FLT3 emerge as additional targets that could contribute to the compounds’ efficacy by reducing cell viability and invasiveness (for instance, PTK6 is linked to migration and its inhibition might reduce metastatic potential). By correlating in silico predictions with in vitro observations, we have built a cohesive rationale for the multifaceted anti-cancer action of Kim-111 and Kim-161. These insights not only validate the compounds’ mechanisms but also guide future development suggesting that further target validation (especially BCL-2) and structure-based optimization could eventually lead to new therapeutic candidates for bladder cancer and possibly other cancers. The computational approach applied here, integrating docking, induced fit refinement, MM-GBSA scoring, and long-timescale MD, exemplifies how in silico modeling can profoundly inform and accelerate drug discovery efforts in identifying and characterizing drug-target interactions in the absence of exhaustive experimental structural biology.