Socio-demographic characteristics of the study participants
Of the 401 patients enrolled in the study, more than half (51.1%) were male participants, with a mean age of 49 years (standard deviation (SD) ± 19 years), and 8.7% of the…
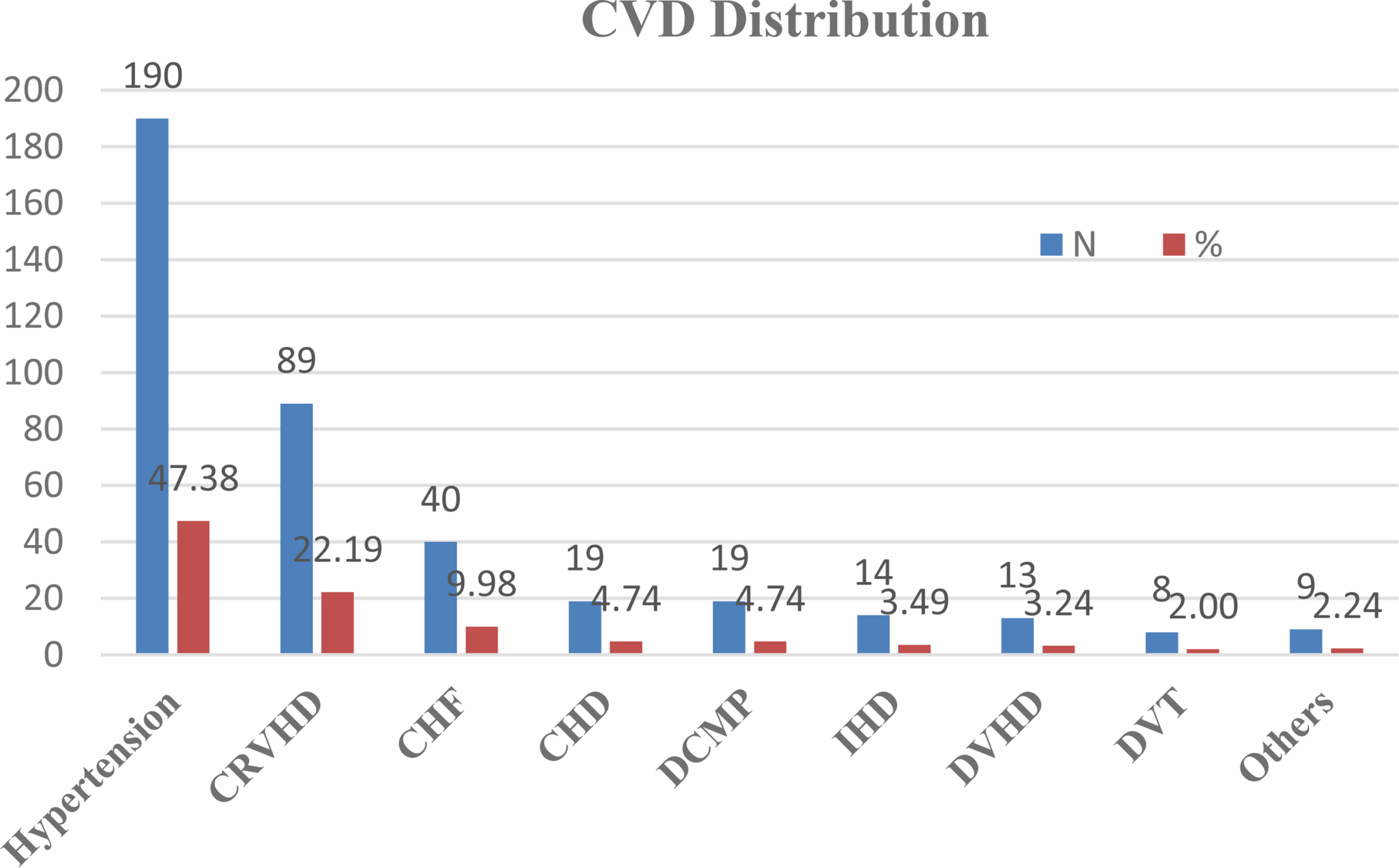
Of the 401 patients enrolled in the study, more than half (51.1%) were male participants, with a mean age of 49 years (standard deviation (SD) ± 19 years), and 8.7% of the…