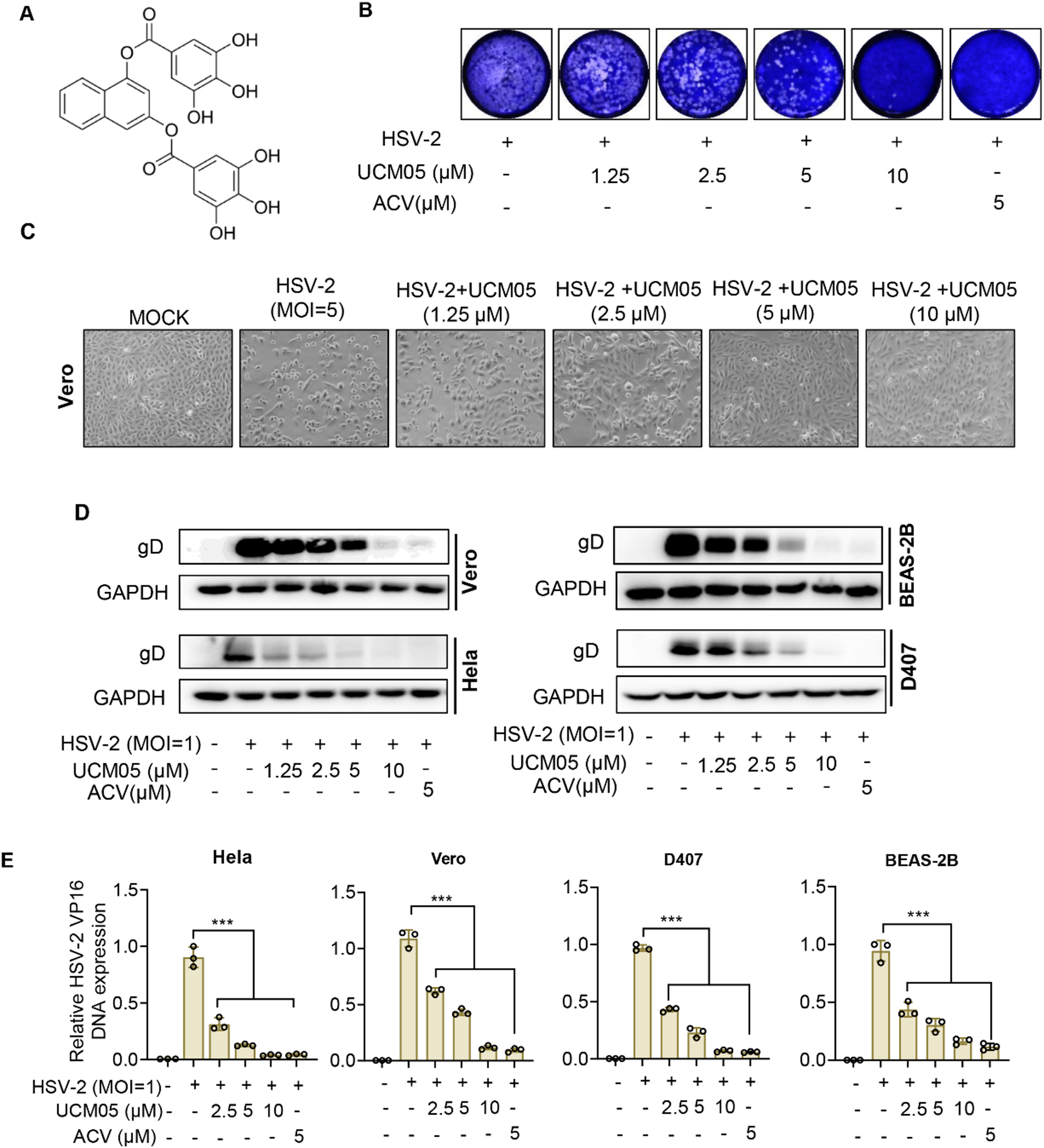
UCM05 effectively suppresses HSV-2 infection while demonstrating low cytotoxicity in vitro
We initially evaluated the anti-HSV-2 activity of UCM05, whose molecular structure is depicted in Fig. 1A, using a plaque reduction assay on Vero cells. As shown in Fig. 1B, the results demonstrate that UCM05 significantly inhibited the production of progeny viruses in a concentration-dependent manner.
The antiviral efficacy of UCM05 against HSV-2 was assessed in vitro. (A) The chemical structure of UCM05. (B) Plaque reduction assay of UCM05 against HSV-2 infection. (C) Cytopathic effect (CPE) in HSV-2 (MOI = 5)-infected Vero cells with different concentrations of UCM05 for 24 h. (D) Immunoblot analysis of HSV-2 glycoprotein D (gD) in HeLa, Beas-2B, Vero and D407 cells infected for 24 h with HSV-2 (MOI = 1) in the presence of the indicated concentrations of UCM05. ACV (5 µM) was used as a positive control. (E) Relative HSV-2 VP16 DNA expression following HSV-2 (MOI = 1) infection of HeLa, Vero, D407 and BEAS-2B cells in the presence of the indicated concentrations of UCM05. Data were shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the control group or indicated by line
The cytotoxicity of UCM05 and ACV on four target cell lines, Vero, HeLa, D407 and Beas-2B, was assessed using MTT assays (Table 1), yeilding with half-maximal cytotoxic concentration (CC50) values between 32.58 ± 0.49 µM and 88.88 ± 2.2 µM. Additionally, the impact of UCM05 on HSV-2 replication was evaluated across the four target cell lines using absolute copy number quantification by RT-qPCR. As detailed in Table 1, UCM05 considerably reduced HSV-2 viral DNA copy numbers in infected cells, with half-maximal inhibitory concentration (IC50) values ranging from 1.57 ± 0.6 to 3.67 ± 0.24 µM. The Treatment Index (TI), calculated as CC50/IC50, further underscores UCM05’s selective antiviral action, with TIs exceeding 20 in all cases, indicating a high degree of specificity and safety for therapeutic use. Notably, UCM05 demonstrated superior inhibitory activity against ACV-resistant HSV-2 strains, with IC50 values considerably lower than those for ACV, highlighting its potential as an effective alternative in resistant cases. For instance, in Hela cells, UCM05’s IC50 against an ACV-resistant strain was 1.03 ± 0.3 µM compared to ACV’s 73.09 ± 1.04 µM. To validate these antiviral effects, optical microscopy-based phenotypic analysis of Vero cells revealed that cells treated with higher UCM05 concentrations (5 and 10 µM) exhibited a significantly diminished cytopathic effect (CPE) caused by HSV-2 (MOI = 5) for 24 h compared to untreated cells or those treated with low concentrations of UCM05 (1.25 µM and 2.5 µM) (Fig. 1C). This result confirms the protective effect of UCM05 on host cells from a morphological perspective of cytopathic effects and further highlights its concentration-dependent antiviral activity. Furthermore, we assessed the expression trends of the HSV-2 envelope protein gD via Western blot analysis. The four target cells were infected with HSV-2 (MOI = 1) and treated with varying concentrations of UCM05 (ranging from 1.25 µM to 10 µM) for 24 h. As shown in Fig. 1D, UCM05 led to a concentration-dependent decrease in gD protein levels across all four tested cell lines. Additionally, absolute copy number quantification by RT-qPCR confirmed a dose-dependent reduction in HSV-2 viral DNA in these cells (P < 0.001; Fig. 1E). These results collectively support the potent antiviral activity of UCM05 by suppressing both viral protein expression and DNA replication.
UCM05 in inhibiting HSV-2 across different stages of viral infection
To determine which stage of the HSV-2 viral life cycle is most sensitive to UCM05, we performed time-of-addition assays under five distinct treatment conditions (Fig. 2A): virus pretreatment, pretreatment of cells (-1 h–0 h), during treatment (0–1 h), post-treatment (1–24 h) of infected cells and entire treatment (0–24 h). Virus pretreatment reflected the viricidal activity of UCM05, while the remaining conditions tested its effects on host-cell interactions and viral replication. The antiviral activity of UCM05 following pretreatment-rinsing may be attributed to its lipophilic-mediated intracellular accumulation, which could directly inactivate virions or prime host antiviral defenses via metabolic/immune regulation, as shown in Fig. 2B and D, UCM05 decreased the expression of gD protein (Fig. 2B), suppressed viral progeny production (Fig. 2C), and reduced viral DNA copy numbers via RT-qPCR (Fig. 2D) across all five conditions (P < 0.001).
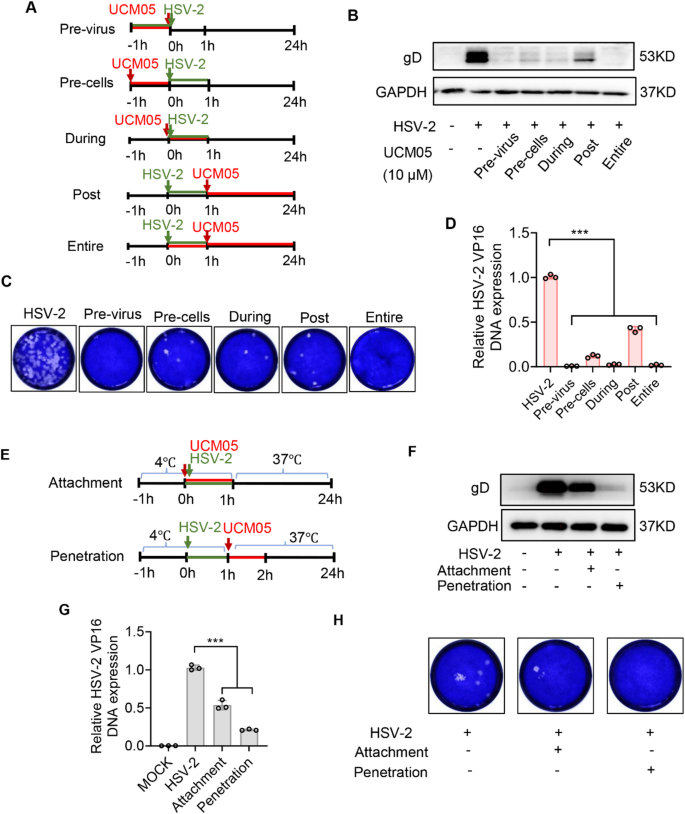
The impact of varied UCM05 treatment scenarios on HSV-2 to infection. (A) UCM05 (10 µM) were added before, during or after the infection of HSV-2 (MOI = 1), respectively as the timeline shown. The effects of different UCM05 treatment conditions on gD expression (B) and viral gene DNA copy numbers (D) were determined by Western blot and RT-qPCR. (C) Different UCM05 treatment conditions against HSV-2 infection were analyzed by plaque reduction assay. (E) Schematic diagram of UCM05 treatment on the attachment and penetration phases of HSV-2 infection. (F) The influence of UCM05 on the attachment and penetration stages of HSV-2 (MOI = 10) infection was investigated using Western blot and (G) RT-qPCR analysis. (H) UCM05 treatment in attachment and penetration phases of HSV-2 infection were evaluated through plaque reduction assay. Data were shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the control group or indicated by line
Considering that treatment remains in the well even after post-infection application, we further investigated whether UCM05 could exert intracellular antiviral effects. To isolate this effect, Vero cells were infected with HSV-2 (MOI = 0.01) for 1 h, after which the viral inoculum was removed and the cells were treated with UCM05 (10 µM) for 8 h. Subsequently, the medium was replaced with 1% methylcellulose overlay to block secondary spread for 72 h. As shown in Figure S1, UCM05 (10 µM) reduced plaque formation by approximately 40% (P < 0.001), indicating its intracellular antiviral activity.
For a more detailed investigation into the specific entry step of HSV-2 that UCM05 inhibits, we analyzed the two distinct phases of virus entry: attachment and post-attachment, the latter also referred to as penetration. As shown in Fig. 2E, virus attachment was evaluated using prechilled Vero cells exposed to 10 µM UCM05 and concurrently infected with HSV-2 (MOI = 10) at 4 °C for 1 h, which promoted virus particle binding to cells but impeded subsequent viral entry stages [31]. Subsequently, we assessed the effect of UCM05 on viral penetration by introducing it to Vero cells at 37 °C for 1 h after HSV-2 infection at 4 °C. After 24 h, Western blot analysis showed a decreasing trend in gD protein expression, and quantitative RT-qPCR revealed reduced viral DNA copy numbers in both phases compared to control (P < 0.001) (Fig. 2F and G). UCM05 reduced the expression of viral protein gD and viral DNA copy numbers (P < 0.001) in attached or penetrated viruses compared to the control group (Fig. 2F and G). Consistent with the expectations, the plaque reduction assay validated these findings (Fig. 2H). Moreover, we observed that UCM05 exerts a more pronounced effect on inhibiting viral penetration. Collectively, these data demonstrate that UCM05 acts at multiple stages of HSV-2 infection, including extracellular virion inactivation, early virus-cell interactions, and intracellular replication, with particular potency in impairing viral penetration.
UCM05 destroyed viral envelope and interacted with viral glycoproteins
Transmission electron microscopy (TEM) was used to observe the structural changes in HSV-2 viral particles upon treatment with UCM05. In control samples, HSV-2 maintained a complete spherical envelope, whereas 100 µM UCM05 treatment led to substantial disintegration of 71.4% of viral envelopes, presenting as fragmented and irregular shapes (Fig. 3A). Molecular docking analyses indicated that UCM05 exhibits strong affinity for HSV-2 glycoproteins gB and gD, with binding energies of -10.1 and − 10.3 kcal/mol, respectively. These interactions suggest a potential mechanism where UCM05 might inhibit viral entry and fusion by altering the conformation of these glycoproteins (Fig. 3B and C).
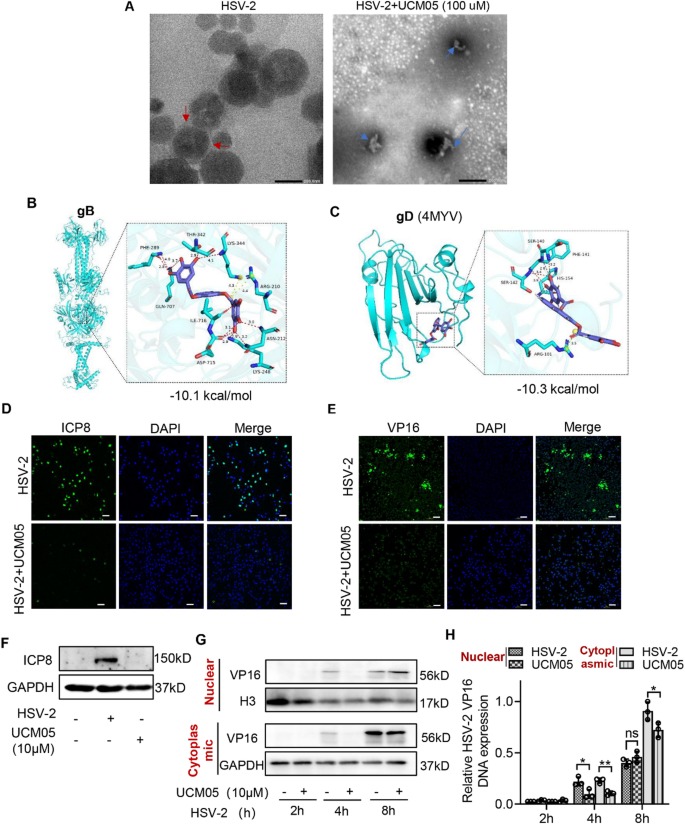
UCM05’s disruption of the HSV-2 viral envelope and its interactions with glycoproteins gB and gD, along with the suppression of HSV-2 viral protein synthesis. (A) The intact spherical structure and envelope of HSV-2 viral particles are highlighted with red arrows on the left, while the right panel illustrates the UCM05-mediated disruption of the HSV-1 envelope (blue arrows). Scale bar, 200 nm. (B) Docking analysis showing the best binding mode between UCM05 and glycoprotein gB. (C) Docking analysis showing the best binding mode between UCM05 and glycoprotein gD. After the supernatant containing HSV-2 was removed, UCM05 was then added to the cells. Localization and expression of (D) ICP8 and (E) VP16 in HSV-2 (MOI = 10) infected D407 cells at 10 hpi measured by confocal microscopy. Scale bar, 100 μm. (F) Western blotting assessing the impact of UCM05 on ICP8 expression in HSV-2-infected (MOI = 10) D407 cells at 10 hpi. (G) The temporal expression of VP16 protein and relative DNA copy numbers in nuclear/cytoplasmic compartments were analyzed by Western blot and RT-qPCR in UCM05-treated D407 cells at 2, 4, and 8 h post-HSV-2 (MOI = 10) infection. Data are presented as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001 compared to the control group or as indicated by lines
Building on our previous findings that UCM05 may counteract HSV-2 infection both intracellularly and extracellularly, we further investigated its effects on key viral proteins involved in HSV-2 replication, specifically VP16 and ICP8. VP16, acting as a potent transcription factor, activates immediate-early gene expression in HSV, which is crucial for initiating the viral replication cycle. Additionally, as a viral capsid protein, VP16 also regulates viral assembly and maturation [32]. ICP8 is the primary single-stranded DNA-binding protein of HSV and possesses helicase activity, playing an essential role in DNA replication [33]. Here, we added UCM05 (10 µM) to D407cells following the removal of the supernatant containing HSV-2 (MOI = 10) for 10 h and observed a concentration-dependent reduction in the expression of the viral proteins ICP8 and VP16 in UCM05-treated groups compared to HSV-2-infected controls using confocal microscopy (Fig. 3D and E). As expected, the Western blot assay confirmed UCM05 inhibited the synthesis of the ICP8 protein (Fig. 3F). We then assessed the impact of UCM05 (10 µM) on VP16 nuclear translocation at distinct intervals post-HSV-2 infection (MOI = 10). Western blot analysis revealed that starting from 4 h post-infection, the expression levels of VP16 in both the nucleus and cytoplasm were reduced in UCM05-treated D407 cells compared to HSV-2-infected cells (Fig. 3G). RT-qPCR analysis showed that at 4 h post-infection, UCM05 primarily reduced the DNA copy number of VP16 gene fragment in the nuclear (P = 0.0281) and cytoplasm (P = 0.0032). At 8 h post-infection, VP16 DNA copy numbers were significantly reduced mainly in the cytoplasm (P = 0.0488) (Fig. 3F). These results suggested that UCM05 treatment inhibited the early nuclear translocation of VP16 to reduce viral replication.
UCM05 treatment protected mice from HSV-2 infection
Next, we evaluated the potential of UCM05 as an antiviral agent in a murine model of genital herpes induced by HSV-2. Mice were administered intraperitoneally with either a placebo (PBS) to the control group and the vehicle group, low-dose UCM05 (15 mg/kg/day), high-dose UCM05 (30 mg/kg/day), or acyclovir (ACV, 100 mg/kg/day) for six consecutive days (Fig. 4A). Although survival was modestly extended in the low-dose UCM05 group, there was no significant improvement in survival rates. Conversely, high-dose UCM05 treatment substantially enhanced survival and provided effective protection against HSV-2, in stark contrast to the complete mortality observed in the HSV-2 infected control group by day 8 post-infection (dpi) (Fig. 4B).
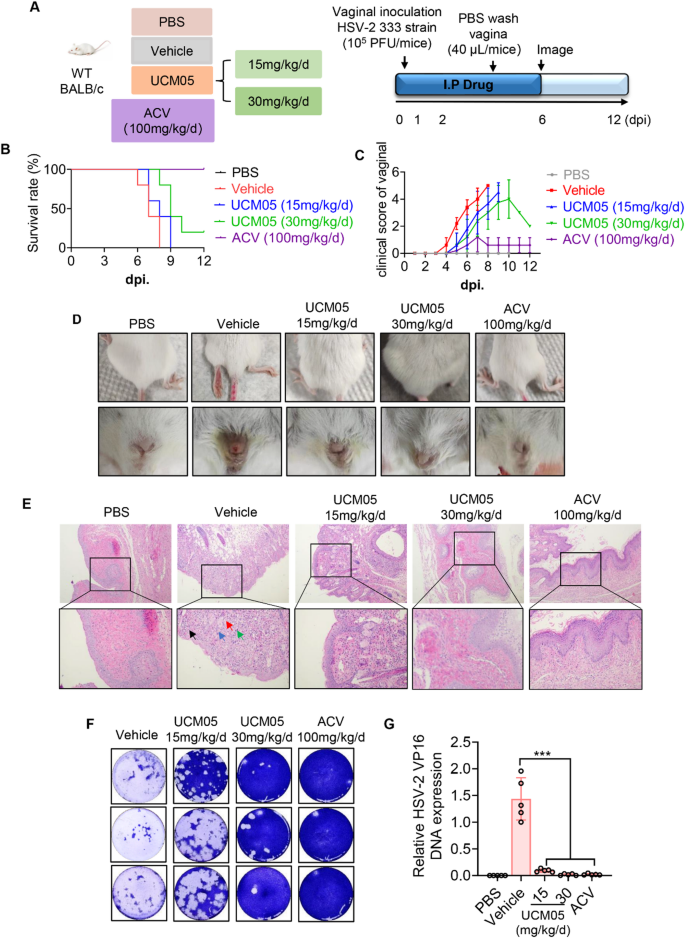
UCM05 treatment protected mice from HSV-2 infections. (A) Evaluation schedule of UCM05 in HSV-2 infected mice therapeutic efficacy. Briefly, following vaginal inoculation with 105 PFU HSV-2 (333 strain) or PBS, BALB/c mice were treated intraperitoneally daily with UCM05 (15 mg/kg/d, 30 mg/kg/d) for 6 days. The control group received an equal volume of PBS as the virus – inoculated group. (B) Survival curve and (C) clinical disease scores (0–5, 5 being severe) of experimental mice in different groups (n = 5) were monitored daily for 12 days post-infection. (D) The representative picture of and the vulva and hindlimb and genital tract stained with hematoxylin-eosin (H&E), genital impairment of experimental mice in different groups at 6 dpi. (E) The virus titers in vaginal washing fluid at 4 dpi were performed by plaque assay (n = 3 per group). (F) RT-qPCR analysis of relative HSV-2 VP16 DNA expression in vagina tissue of experimental mice in different groups. Data were shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the control group or indicated by line
Consistent with these observations, UCM05 treatment significantly reduced the clinical scores of acute vaginal injuries compared to the HSV-2 infected group, as shown in Fig. 4C. Figure 4D depicts that by day 6 post-infection (dpi), the HSV-2 infected group exhibited a significant worsening of progressive vulvar erosion and hindlimb paralysis. In contrast, these symptoms were markedly alleviated in both the UCM05-treated and ACV-treated groups. Additionally, the effect of UCM05 on HSV-2 infection in vivo was further substantiated by histopathological analysis of the genital tract at 6 dpi (Fig. 4E). Necrotic areas (indicated by blue arrows), neutrophils (green arrows), inflammatory debris (black arrows), and viral inclusion bodies (red arrows) were observed in the vaginal submucosa and adipose tissue of HSV-2-infected mice. Notably, these pathological changes were notably diminished following treatment with UCM05 and ACV, Consequently, UCM05 significantly reduced reproductive tract damage and the inflammatory response in HSV-2-infected mice.
To further assess the impact of UCM05 on HSV-2 replication in vivo, the viral titers in vaginal lavage were quantified at 4 dpi using plaque assays. Results showed that the viral loads of vaginal lavage significantly decreased in the UCM05 treatment groups in a concentration-dependent manner (Fig. 4F). Moreover, UCM05 treatment resulted in a significant reduction in HSV-2 viral DNA copy numbers (P < 0.001) in the vagina compared to the HSV-2-infected group, as shown in Fig. 4G. These results indicate that UCM05 effectively suppresses HSV-2 replication in vivo.
UCM05 reduces HSV-2 replication by inhibiting fatty acid synthesis
UCM05 has been identified as an effective inhibitor of FASN, a key enzyme in the de novo lipogenesis pathway, catalyzing the synthesis of long-chain fatty acids from acetyl-CoA and malonyl-CoA [34]. The process is regulated by the activation of sterol regulatory element-binding protein 1 (SREBP1), a transcription factor that upregulates the expression of FASN [35], as well as other lipogenic genes such as stearoyl-CoA desaturase 1 (SCD1) and acetyl-CoA carboxylase (ACC1). SCD1 introduces a double bond into saturated fatty acids, converting them into monounsaturated fatty acids [36]. ACC1 is involved in the initial step of fatty acid synthesis, producing malonyl-CoA from acetyl-CoA, which is then utilized by FASN [37]. These enzymes constitute a coordinated regulatory network that controls the synthesis and metabolism of fatty acids within the cell. Previous studies have demonstrated that multiple viruses, including SARS-CoV-2, hepatitis C virus (HCV), Singapore Irivirus (SGIV) and Porcine Reproductive and Respiratory Syndrome Virus (PRRSV), rely on fatty acid synthesis for their replication [37,38,39]. To investigate whether HSV-2 infection affects cellular fatty acid metabolic pathway, we conducted a Gene Set Enrichment Analysis (GSEA) using data from relevant databases (Fig. 5A). The results revealed that the lipid metabolism pathway was significantly downregulated in HSV-2 infected genital epithelial lesion compared to contra-lateral normal skin biopsies. RT-qPCR analysis demonstrated that HSV-2 infection downregulated the mRNA expression of key fatty acid metabolism genes (SCD1, SREBP1, ACC1) in a time- and dose-dependent manner. Specifically, at 36 h post-HSV-2-infection and a MOI of 5, the expression levels of these genes (SCD1, P < 0.001; SREBP1, P < 0.001; ACC1, P = 0.0215 (MOI = 1, 36 h); ACC1, P = 0.0331 (MOI = 5, 24 h)) significantly decreased (Fig. 5C). Immunofluorescence and Western blot assays demonstrated a reduction trend in FASN and SREBP1 expression levels in HSV-2-infected (MOI = 5) D407 cells for 24 h compared to uninfected controls (Fig. 5B and D). Additionally, both extracellular (P = 0.0293) and intracellular (P = 0.0199) free fatty acid levels were reduced in HSV-2-infected cells (MOI = 5) for 24 h, underscoring that HSV-2 infection regulates de novo fatty acid synthesis in D407 cells (Fig. 5E).
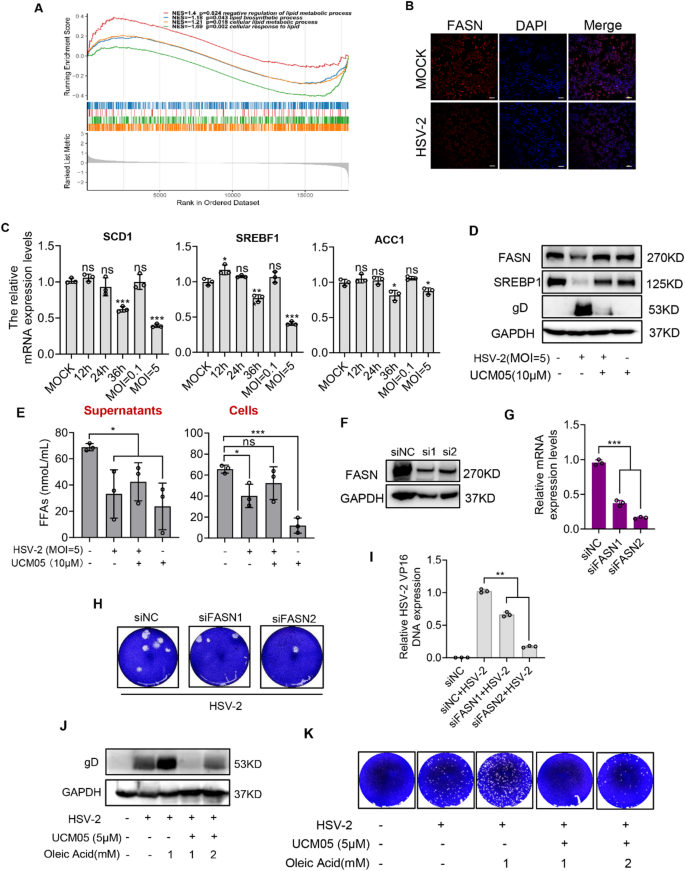
UCM05 reduces HSV-2 replication by inhibiting fatty acid synthesis. (A) GSEA of differentially expressed genes in HSV-2 infected genital epithelial lesion versus contra-lateral normal skin biopsies. Gene sets were derived from the The Gene Expression Omnibus (GEO) database (GSE172423). The analysis was performed using the weighted enrichment score algorithm, with a threshold for significance set at p-value < 0.05 and false discovery rate (FDR) q-value < 0.25. The figure displays four significantly enriched gene sets related to lipid metabolism, including Normalized Enrichment Scores (NES) and nominal p-values. (B) Fluorescence labeling of FASN (red) in D407 cells, with DAPI labeled nuclei (blue), following a 24-hour HSV-2 (MOI = 5) infection. Scale bars represent 100 μm. (C) Relative mRNA expression of SCD1, SREBF1, and ACC1 in D407 cells infected with HSV-2, normalized to GAPDH. (D) Western blot analysis of FASN and SREBF1 protein expression in D407 cells infected with HSV-2 (MOI = 5) after a 24-hour treatment with or without UCM05 (10 µM). (E) D407 cells were infected with HSV-2 (MOI = 5) for 24 h. The precipitation was removed after centrifugation, and the cell supernatant was collected for detection of FFAs content by colorimetric assay. (F) Immunoblot analysis and (G) mRNA expression of FASN in D407 cells transfected for 48 h with non-targeting control siRNA (siNC) or siRNA targeting FASN (si1 or si2). (H) Plaque reduction assay of FASN knockdown on HSV-2 infection. (I) The DNA copy numbers of viral genes were analyzed by RT-qPCR. (J) Adding oleic acid (OA) or UCM05 to HSV-2-infected and uninfected D407 cells. After incubation for 24 h, the cells were collected for gD protein expression analysis by Western blot, (K) the supernatant of cells was harvested for virus titration by plaque assay. Data were shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the control group or indicated by line
To further elucidate the role of UCM05, we infected D407 cells with HSV-2 in the presence or absence of UCM05 (10 µM). After 24 h, we collected the cells and supernatants for Western blot analysis and free fatty acid (FFA) quantification. We found that, even without HSV-2 infection, UCM05 slightly reduced the protein levels of FASN and SREBF1 and lowered both extracellular (P = 0.0123) and intracellular (P < 0.001) FFA content. Moreover, under HSV-2 infection, UCM05 reversed the virus-induced downregulation of FASN and SREBF1 protein levels, as well as the decreased FFA content in the supernatant (P = 0.0362) (Fig. 5D and E), which indicated that UCM05 alleviated the HSV-2-induced fatty acid metabolism abnormalities in host cells.
Subsequently, we transfected D407 cells with two distinct siRNAs targeting FASN and maintained the transfection for 48 h. Both siRNAs effectively knocked down FASN, resulting in a reduction trend in FASN protein levels and a significant decrease in mRNA expression. (P < 0.001) (Fig. 5F and G). HSV-2 infection (MOI = 1) was performed for an additional 24 h after transfection of D407 cells with siRNAs for 24 h. Subsequently, cells and supernatants were collected for plaque assays and RT-qPCR analysis. In the two FASN knockdown group, the progeny virus titers of HSV-2 were significantly reduced, as evidenced by decreased plaque formation (Fig. 5H). The results of RT-qPCR analyses showed that partial depletion of FASN by siRNA significantly reduced the HSV-2 viral DNA copy numbers (P < 0.001), indicating that FASN activity supports the replication of HSV-2 (Fig. 5I).
Furthermore, we conducted rescue experiments by adding oleic acid (OA) or UCM05 to HSV-2-infected and uninfected D407 cells. We observed that adding OA increased the levels of viral protein gD and progeny virus production compared to conditions without OA. While UCM05 (5 µM) significantly reduced viral protein levels and virus yield, increasing the OA concentration from 1 mM to 2 mM weakened the inhibitory effect of UCM05 (Fig. 5J and K). These results suggested that the increase in fatty acids could promote HSV-2 replication, while UCM05 reduced viral replication by inhibiting this synthetic process.
Collectively, these findings highlight that UCM05 reduces HSV-2 replication through the inhibition of FASN and fatty acid metabolism, supporting the potential of UCM05 as a therapeutic agent targeting virus-associated lipid metabolism.
Impact of UCM05 on inflammatory responses in HSV-2 infection
Type I interferons (IFN-I) are pivotal mediators of the host immune response during viral infections, exerting broad-spectrum antiviral activities that suppress viral replication [40]. To assess the role of UCM05 in antiviral immunity, we performed RT-qPCR analyses on D407 cells stimulated with HSV-2 (MOI = 10) for 8 h or the DNA virus analogue Poly(dA: dT) (10ng/mL) for 24 h, both with and without UCM05 treatment. While UCM05 treatment led to a reduction in the mRNA levels of IFN-I-related genes in HSV-2-infected cells, but it was uncertain whether this effect was a result of its antiviral activity. To clarify this, we further stimulated cells with poly(dA: dT), which revealed that UCM05 significantly increased the expression of IFN-β and various interferon-stimulated genes (ISGs), including ISG15, ISG54, IFIT1, and IFIT3 (Fig. 6A and B). These findings imply that UCM05 may enhance antiviral immunity by modulating IFN-I production.
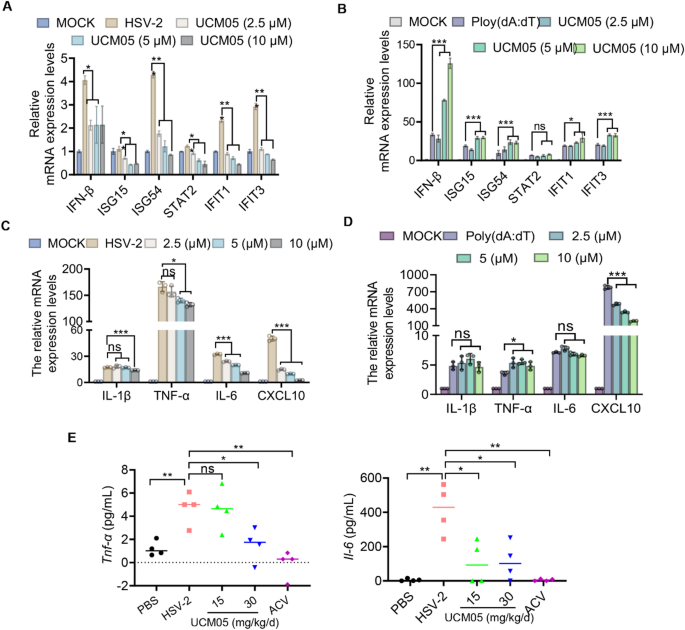
Impact of UCM05 on Inflammatory Responses in HSV-2 infection. (A) The effect of UCM05 on the mRNA expression levels of IFN-I relatied genes (IFN-β, ISG15, ISG54, STAT2, IFIT1, and IFIT3) in HSV-2 infected (MOI = 10) D407 cells treated with various concentrations of UCM05 (2.5, 5, 10 µM) for 8 h. MOCK represents uninfected control. (B) mRNA expression levels of IFN-I related genes in response to synthetic viral mimic Poly(dA: dT) stimulation in the presence of UCM05 at varying concentrations. (C) The effect of UCM05 on the mRNA expression levels of inflammatory cytokines (IL-1β, TNF-α, IL-6, CXCL10) in HSV-2 infected D407 cells treated with various concentrations of UCM05 (2.5, 5, 10 µM). MOCK represents uninfected control. (D) mRNA expression levels of inflammatory cytokines in response to Poly(dA: dT) stimulation in the presence of UCM05 at varying concentrations. (E) Concentration of Tnf-α and Il-6 in the serum of mice treated with UCM05 or acyclovir (ACV) compared to control groups treated with PBS or infected with HSV-2 alone (n = 4) at 6 dpi. Data were shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the control group or indicated by line
In parallel, histopathological assessments indicated that UCM05 treatment could mitigate the inflammatory response induced by HSV-2 infection. We further investigated this by evaluating the impact of UCM05 on the expression of inflammatory cytokines in both in vitro and in vivo models. Specifically, UCM05 significantly downregulated the mRNA expression of key inflammatory cytokines, such as IL-1β, TNF-α, IL-6, and CXCL10, in D407 cells triggered by HSV-2 infection, demonstrating a dose-dependent effect at concentrations of 2.5, 5, and 10 µM (Fig. 6C). UCM05 treatment reduced the mRNA level of CXCL10 stimulated by poly(dA: dT) without leading to increased levels of other inflammatory factors (Fig. 6D). Furthermore, in vivo studies corroborated the anti-inflammatory effects of UCM05, showing that treatment at doses of 15 and 30 mg/kg/day significantly reduced the levels of Tnf-α and Il-6 in the serum of HSV-2-infected mice (n = 4) at 6 dpi (Fig. 6E). These results collectively demonstrate that UCM05 can effectively reduce inflammatory responses associated with HSV-2 infection without causing excessive production of inflammatory cytokines.
UCM05 effectively inhibits HIV-1, both HIV-1/HSV-2 coinfections and HSV-1 infections
Our comprehensive analysis demonstrated that UCM05 significantly inhibits the replication of various viruses, including HIV-1, both HIV-1/HSV-2 coinfections and HSV-1. We evaluated the cytotoxicity of UCM05 on TZM-bl cells across a therapeutic concentration range of 5–20 µM. Within this range, no more than 20% of cell viability was reduced in TZM-bl cell line (Figure S2). We added HIV-1SF162 virus (MOI = 1) to pre-plated TZM-Bl cells, in the presence or absence of UCM05 (5–20 µM). The cells were incubated at 37 °C for 48 h. The inhibitory effect of UCM05 on HIV-1SF162 proliferation was evaluated using a luciferase reporter system. In TZM-bl cells infected with replication-competent HIV-1SF162 (MOI = 1) and co-infected with HIV-1SF162 (MOI = 1) and HSV-2 (MOI = 0.5) for 48 h, UCM05 reduced the replication of HIV-1SF162 in a dose-dependent manner(P < 0.001) (Fig. 7A). The compound’s inhibitory effects on co-infection were further confirmed through plaque reduction assays, showing a clear decrease in the number of HSV-2 viral plaques with increasing concentrations of UCM05 from 5 to 20 µM (Fig. 7B). Additionally, Western blot analysis corroborated these findings, revealing a trend of reduction in the levels of HSV-2 gD protein across various concentrations of UCM05 in the HIV-1/HSV-2 co-infected samples (Fig. 7C).
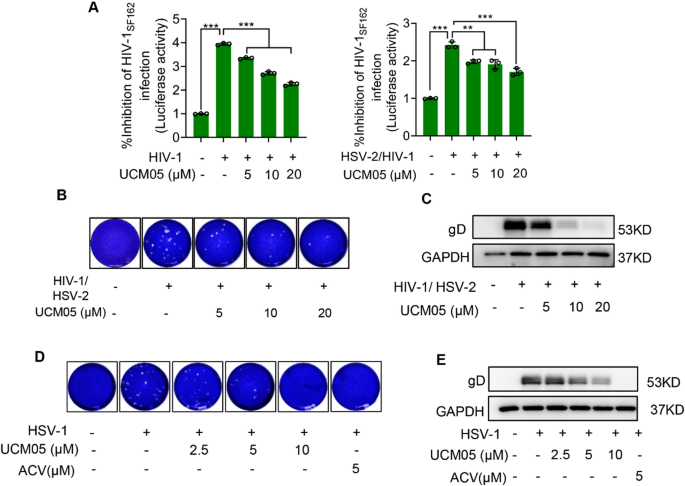
UCM05 inhibits both HIV/HSV-2 co-infection and HSV-1. (A) TZM-bl cells were infected with replication-competent HIV-1SF162 (MOI = 1) with or without HSV-2 (MOI = 0.5) co-infection, then treated with indicated concentrations of UCM05 for 48 h. Viral replication was quantified through luciferase reporter activity. (B) The inhibitory effects of UCM05 treatment on HIV-1/HSV-2 co-infection were analyzed by a plaque reduction assay and (C) Western blot analysis. (D) Plaque reduction assay and (E) Immunoblot analysis of the indicated concentrations of UCM05 against HSV-1 (MOI = 1) infection for 24 h in Vero cells. Data were shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the control group or indicated by line
For HSV-1, similar dose-dependent inhibitory effects were observed. Plaque assays conducted on HSV-1 infected samples highlighted a marked reduction in plaque formation as UCM05 concentrations increased (Fig. 7D). This was paralleled by a decrease in gD protein levels, as shown by immunoblot analysis (Fig. 7E), underscoring the antiviral efficacy of UCM05 against both types of herpes simplex virus. These results collectively establish UCM05 as a potent inhibitor against multiple viral infections, such as HIV-1, HIV-1/HSV-2 coinfections and HSV-1.