Clinical presentation and disease progression
A 42-year-old female power plant worker was admitted to our respiratory department on August 1, 2021, due to a recurrent cough and sputum production that had persisted for over eight years and worsened in the past week. One week prior to admission, following a common cold, the patient experienced a recurrence of symptoms accompanied by a low-grade afternoon fever ranging from 37.5 to 37.8 °C. She reported occasional chest tightness, dyspnea, and palpitations, which were relieved by rest.
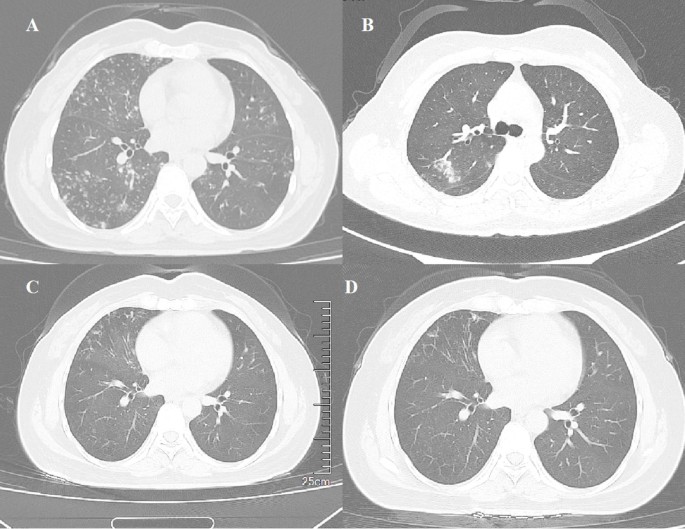
On physical examination, bilateral coarse breath sounds were noted, with a few moist rales in the lower lung fields. Laboratory investigations revealed no significant abnormalities in routine blood tests, C-reactive protein (CRP) and procalcitonin. Initial bacterial smears showed Gram-positive cocci and Gram-negative bacilli, but no acid-fast bacilli were detected. The tuberculosis γ-interferon test was negative. Thyroid function tests revealed elevated thyroid peroxidase antibodies (398.1 U/mL), an erythrocyte sedimentation rate (ESR) of 22 mm/h, and a positive purified protein derivative (PPD) test with a 10 mm induration. Thyroid ultrasound indicated diffuse thyroid disease suggestive of Hashimoto’s thyroiditis. Chest CT demonstrated scattered nodules and high-density spots in both lungs, along with traction bronchiectasis in the right middle lobe (Fig. 1A).
Views of computed tomographic scan of the chest at different stages of disease. Chest computed tomography (CT) scans show multiple centrilobular nodules in both lungs, with some presenting a tree-in-bud appearance and some presenting as patchy ground-glass opacities, and traction bronchiectasis in the middle lobe of the right lung on July 26, 2021 (A); new patchy ground-glass opacity in the upper lobe of the right lung on April 3, 2023 (B); multiple centrilobular nodules in both lower lungs on July 28, 2023, with significant reduction compared to previous images (C); multiple centrilobular nodules in both lower lungs further reduced and absorbed, along with a decrease in traction bronchiectasis in the right middle lobe on June 18, 2024 (D).
The patient was treated with clarithromycin (1,000 mg every other day), rifampin (600 mg every other day), and ethambutol (750 mg every other day), as the mNGS on August 6, 2021 (refer to the mNGS analysis section) indicated the presence of NTM. The treatment continued for 17 months. In April 2023, chest CT revealed new patchy ground-glass opacities in the right upper lobe and increased bilateral lung inflammation (Fig. 1B). T cell subsets in the blood showed increased helper T lymphocytes (45.14%) and decreased cytotoxic T lymphocytes (12.93%), with the absolute count of cytotoxic T lymphocytes being 258.69 cells/µL. CD3 + CD4+/CD3 + CD8 + ratio was 3.49 (normal 0.7–2.8). Considering the absence of significant clinical improvement after a 17-month antibiotic regimen, and in light of recommendations from clinical practice guidelines of leading international respiratory medicine and infectious diseases societies39 as well as empirical regimens for Mycobacterium paraffinicum40the treatment was subsequently was adjusted to include amikacin (0.4 g daily, IV), azithromycin (0.5 g daily), ciprofloxacin (1,000 mg daily), and linezolid (600 mg twice daily). By July 28, 2023, the patient’s symptoms had improved, with significant absorption of lung lesions on chest CT, though right middle lobe bronchiectasis persisted (Fig. 1C). The treatment regimen was continued with azithromycin (0.5 g daily), ciprofloxacin (1,000 mg daily), and linezolid (300 mg twice daily), which has been ongoing for a year. The most recent CT scan on June 18, 2024, showed further reduction and absorption of multiple centrilobular nodules in both lower lungs, along with a reduction in traction bronchiectasis in the right middle lobe (Fig. 1D).
Metagenomic next-generation sequencing analysis
We performed three rounds of mNGS, with total nucleic acids for each analysis independently extracted from freshly collected BALF samples at the respective time points. In the initial round of mNGS of BALF (SRA accession number SRR30415380) conducted on August 6, 2021, we detected one sequence attributed to Mycobacterium intracellulare (M. intracellulare). It is crucial to note that this sequence is not exclusive to M. intracellulare but is also common among other species of NTM. A repeat BALF mNGS on April 25, 2023, identified Nocardia cyriacigeorgica (171 sequences), Mycobacterium paraffinicum (M. paraffinicum, 41 sequences), and Mycobacterium tuberculosis complex (2 sequences). Although a relatively high sequence count of Nocardia cyriacigeorgica was detected, metagenomic sequencing cannot distinguish colonization from infection. Given that Nocardia species are common environmental saprophytes and transient colonizers in immunocompromised hosts, combined with the absence of typical nocardiosis symptoms or radiological features and the patient’s improvement with NTM-targeted therapy alone, we interpreted its detection as incidental colonization rather than active infection. Notably, a total of 676 sequences were classified at the genus level as Mycobacterium, suggesting the potential presence of a novel species that could not be confidently assigned to any known species (SRA accession number SRR30415381). On June 20, 2023, another mNGS of BALF detected M. paraffinicum (110 sequences), while 1,181 sequences were identified at the genus level as Mycobacterium (SRA accession number SRR30415382). The classification of some sequences as M. paraffinicum suggests that this potential new NTM species may be highly similar to M. paraffinicum, further complicating precise species-level identification.
Results of rapid genetic detection and bacterial isolation and characterization
Rapid genetic detection of TB/NTM infections and screening for drug resistance genes, conducted on BALF using the DNA microarray method, also confirmed the presence of Mycobacterium species, but Mycobacterium tuberculosis was not detected. Additionally, no resistance genes for isoniazid or rifampin were identified. A mycobacterial strain was successfully isolated from BALF and subsequently subjected to further characterization on July 13, 2023. The Mycobacterium culture showed growth of smooth colonies with yellow pigmentation on Löwenstein-Jensen (LJ) medium regardless of light exposure, indicative of carotenoid production, a characteristic feature of certain scotochromogenic NTM species (Fig. 2A). Acid-fast staining was positive, revealing pink or red rod-shaped bacilli (Fig. 2B).
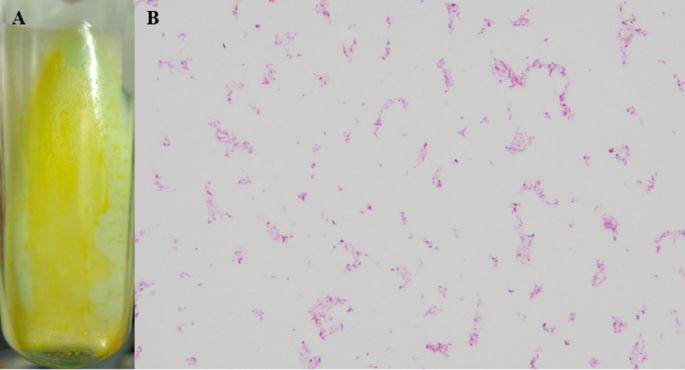
Colonial morphology (A) and acid-fast staining property (B) of Mycobacterium hainanense. (A) Yellow-pigmented colonies of Mycobacterium hainanense HNNTM2301 grown on Löwenstein-Jensen medium. The colonies exhibit characteristic slow growth and pigmentation typical of nontuberculous mycobacteria (NTM). (B) Acid-fast staining of HNNTM2301 reveals typical pink or red rod-shaped bacteria. This staining method confirms the presence of mycobacteria due to their mycolic acid-rich cell walls, which retain the dye.
Phylogenetic analysis, DNA-DNA hybridization and average nucleotide identity
The whole genome sequencing (WGS) of the isolated strain HNNTM2301 was conducted, and the raw sequencing reads and assembled genome were uploaded to the NCBI SRA database (accession number SRR33114765 and SRR33114766) and RefSeq database (accession number GCF_041890355.1) respectively. The values of digital DNA-DNA hybridization (dDDH) and average nucleotide identity (ANI), along with phylogenetic analysis, were then used to compare our isolated strain with 103 representative genomes of the genus Mycobacterium in the RefSeq database to confirm the species (Fig. 3). The whole-genome phylogenetic tree grouped our strain with M. paraffinicum and Mycobacterium nebraskense (M. nebraskense) in the same subcluster. Pairwise comparisons showed dDDH (d4) and ANI values between our strain and the representative genomes of Mycobacterium, with both the highest values observed for M. nebraskense (accession number GCF_001021495.1): 34.3% for dDDH and 88.07% for ANI (Fig. 3). These values are below the thresholds of 70% for dDDH and 95–96% for ANI, which are used for bacterial species delineation32,41. The dDDH and ANI values between strain HNNTM2301 and the remaining closest five type strains M. paraffinicum, M. seoulense, M. parascrofulaceum, M. paraseoulense, and M. scrofulaceum, were all approximately 33.3% and 87.8%, respectively (Fig. 3). A further ANI comparison of our strain with a total of 8,139 Mycobacterium genomes available in the RefSeq database showed the highest value to M. paraffinicum (accession number GCF_001907675.1) and M. scrofulaceum (accession number GCF_001667885.1), with similarities of 92.06% and 91.74%, respectively.
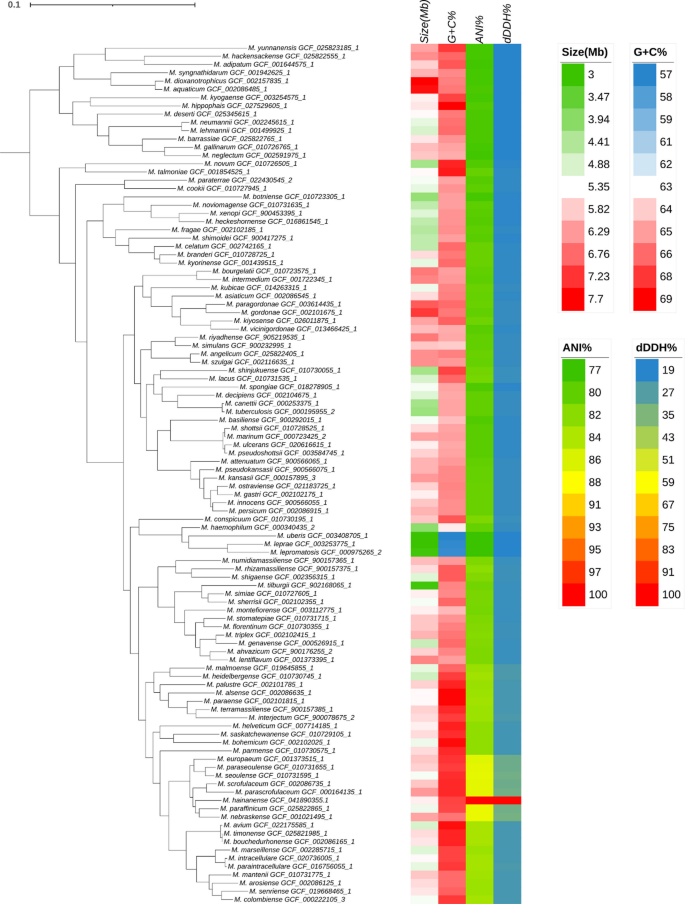
Phylogenetic tree and pairwise comparisons of genome size, GC content, dDDH (d4) and ANI values between Mycobacterium hainanense HNNTM2301 and type strains of Mycobacterium. The phylogenetic tree was inferred using EasyCGTree software based on 120 single-copy protein-coding genes and rooted at the midpoint. The strain we isolated in this study was named Mycobacterium hainanense. The genomes of 103 type strains of Mycobacterium were downloaded from the RefSeq database accessed on March 26, 2024. The maximum-likelihood phylogeny shows the genome size, GC content, and pairwise comparisons of dDDH (d4) and ANI values between M. hainanense and other Mycobacterium species.
Identification of isolates by multilocus analysis
The gene sequences of 16 S rRNA (1493 bp), hsp65 (441 bp), rpoB (752 bp) and sodA (464 bp) were aligned separately for strain HNNTM2301 and the 103 reference mycobacterial strains using a multiple alignment algorithm, followed by the construction of phylogenetic trees. The phylogenetic tree based on the 16 S rRNA gene sequences revealed that the isolated strain HNNTM2301 was most closely related to type strain of M. scrofulaceum and M. paraffinicum with a bootstrap value of 80 (Fig. 4A). Additionally, the online BLAST analysis results for the 16 S rRNA of strain HNNTM2301 showed the closest match (99.8%) with M. paraffinicum strain ATCC 12670.
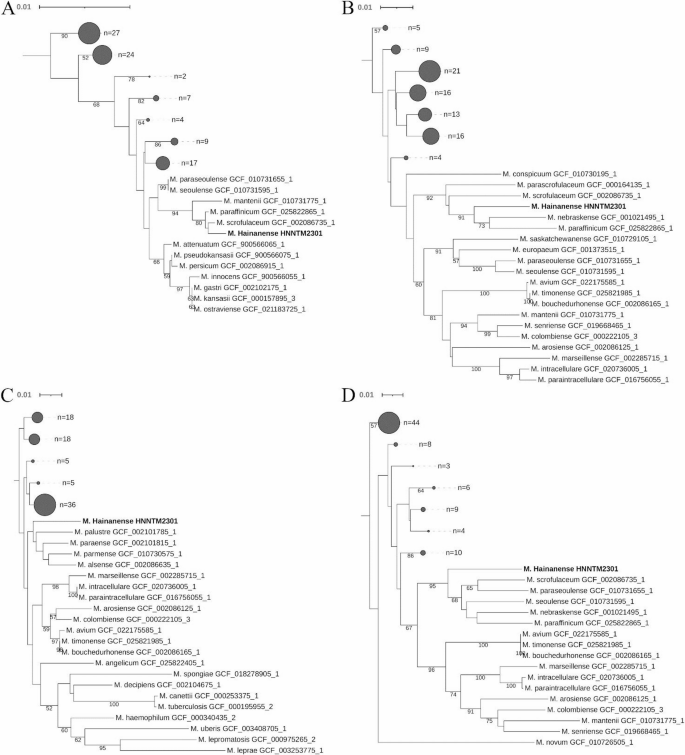
Phylogenetic relationships of strain HNNTM2301 with other species of the genus Mycobacterium based on the 16 S rRNA gene (A), rpoB gene (B), hsp65 gene (C) and sodA gene (D). These trees were reconstructed using the neighbor-joining method with the Kimura 2-parameter distance correction model. Bootstrap values were calculated from 1,000 replications. Bootstrap values below 50% are not shown. Subtrees that are collapsed are represented as filled circles, with the circle size indicating the number of strains in each subtree. The 16 S rRNA gene was not detected in the genome of the type strain of Mycobacterium uberis (GCF_003408705.1), while the sodA gene was absent in the type strain of Mycobacterium gallinarum (GCF_010726765.1), Mycobacterium barrassiae (GCF_025822765.1) and Mycobacterium neglectum (GCF_002591975.1).
The phylogenetic analysis based on the partial rpoB gene sequences supported the grouping of strain HNNTM2301, M. nebraskense and M. paraffinicum in the rpoB gene-based tree with a bootstrap value of 91 (Fig. 4B). Sequence similarities for rpoB between strain HNNTM2301 and the representative M. nebraskense and M. paraffinicum were 96.54% and 95.48%, respectively.
In the hsp65-sequence-based phylogenetic analysis, strain HNNTM2301 was clustered with M. palustre, M. paraense, M. parmense and M. alsense. However, the bootstrap value of the group was below 50 (Fig. 4C). Sequence similarities for hsp65 between strain HNNTM2301 and type strain of M. palustre, M. paraense, M. parmense and M. alsense were 95.92%, 96.37%, 96.15%, and 96.83%, respectively. Based on a further online BLAST analysis, we found that the highest similarities to HNNTM2301 were with M. scrofulaceum (GenBank: GQ478700.1) and M. parascrofulaceum (GenBank: HM454226.1), at 99.55% and 99.32% respectively.
Also, a phylogenetic tree based on sodA gene sequences revealed that strain HNNTM2301 clustered together with M. scrofulaceum, M. paraseoulense, M. seoulense, M. nebraskense, and M. paraffinicum (Fig. 4D). Gene sequence similarities among these strains showed that the closest phylogenetic relationship was between strain HNNTM2301 and M. seoulense (93.75% sequence similarity).
Taken together, the uniqueness of four independent gene sequences (16 S rRNA, rpoB, hsp65, and sodA) together with the lower DNA-DNA relatedness and whole genomic similarity support the suggestion that strain HNNTM2301 is delineated from M. paraffinicum, M. nebraskense and M. scrofulaceum which are the most closely related species (Table 1). It was concluded that the strain represents a novel species for which the name Mycobacterium hainanense sp. nov. is proposed with type strain HNNTM2301.
Genome characterization and functional analysis
The genome of strain HNNTM2301 was sequenced and assembled into a 5,800,079 bp circular chromosome with a GC content of 67.88% (Fig. 5). The genome contained 5,396 coding sequences, 47 tRNA genes, 3 rRNA genes (including 23 S rRNA, 16 S rRNA, and 5 S rRNA), and 3 ncRNAs genes. The genome sequence and its annotation information were submitted to the NCBI database under accession number NZ_CP169059. Based on the WGS-predicted phenotype, no resistance was detected against streptomycin, amikacin, bedaquiline, ethambutol, isoniazid, rifampicin, or linezolid.
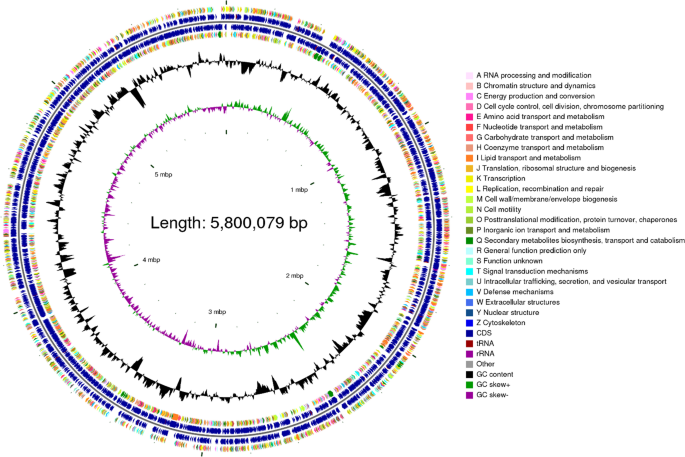
Circular representation of the genome of Mycobacterium hainanense HNNTM2301. This circular genome map comprises six concentric rings: the first and fourth rings represent coding sequences (CDS) on the forward and reverse strands, with colors denoting COG functional categories; the second and third rings display CDS, tRNA, and rRNA genes on the forward and reverse strands; the fifth ring depicts GC content, where outward peaks indicate regions with higher GC content and inward peaks denote lower GC content relative to the genome average; the sixth ring presents GC-Skew values, calculated as (G − C)/(G + C), which reflect strand-specific GC composition.
COG analysis annotated 4,228 genes, categorized into 23 functional groups. The majority of genes were involved in the pathways of lipid transport and metabolism, transcription, coenzyme transport and metabolism, and energy production and conversion (Fig. 6A). A total of 3,869 genes were annotated in the GO database, with the most enriched pathways being the integral component of the membrane (881 genes) and the cytoplasm (275 genes) in cellular components. DNA binding and ATP binding were the most enriched molecular functions, with 359 and 302 genes, respectively. Biological processes related to the regulation of DNA-templated transcription (149 genes) and methylation (104 genes) showed the highest gene counts (Fig. 6B). Furthermore, 2,243 orthologous protein-coding genes were assigned to 43 KEGG metabolic pathways, with the highest gene enrichment observed in global and overview maps, carbohydrate metabolism, amino acid metabolism, energy metabolism and lipid metabolism, which are critical for bacterial metabolism (Fig. 6C). These findings align with the COG metabolic pathway analysis, revealing that many genes contribute to essential bacterial metabolic processes.
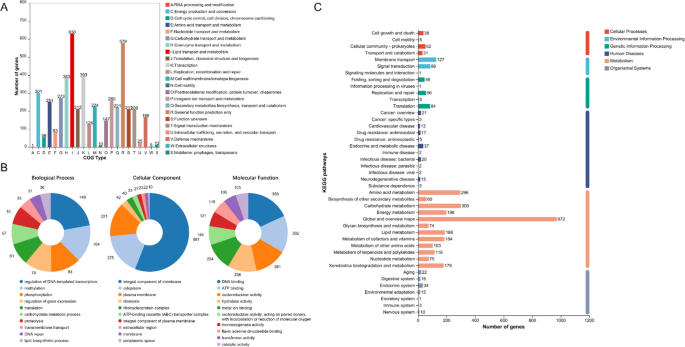
Functional annotation of Mycobacterium hainanense HNNTM2301 based on COG (A), GO (B), and KEGG (C) classifications. (A) COG functional classification of strain HNNTM2301, with 4,228 genes categorized into 23 COG types. (B) GO classification of strain HNNTM2301, with 3,869 genes assigned to 42 subcategories across three primary GO domains. (C) KEGG classification of strain HNNTM2301, with 2,243 genes mapped to 43 KEGG pathways.