FORECAST HIGHLIGHTS
-
SA STAYS JUST ABOVE FREEZING: Most of Bexar County stays above the freezing mark this morning
-
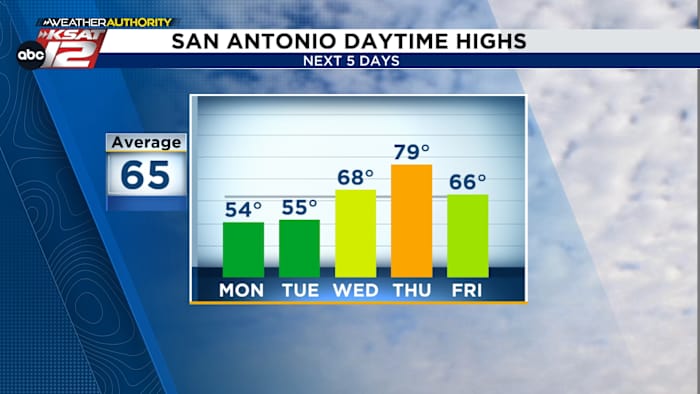
MOSTLY CLOUDY & COOL TODAY: Few peeks of sun, highs in the 50s
-
SMALL RAIN CHANCE TUESDAY: Light showers possible Tuesday afternoon…