Cell culture
U87 and HEK cell lines were cultured at 37 ∘C and 10% CO2 in DMEM D6429 media (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS), 50 μg ml−1 streptomycin and 50 units ml−1 penicillin.
T2 cells and Nalm6 cells were cultured at 37 ∘C and 10% CO2 in RPMI 1640 (Sigma-Aldrich) supplemented with 10% FBS, 50 μg ml−1 streptomycin and 50 units ml−1 penicillin.
All cell lines were obtained from the ATCC except for Nalm6, which was provided by Crystal Mackall.
Primary human T cells were isolated from leucocyte cones and cultured at 37 ∘C and 10% CO2 in RPMI 1640 (Sigma-Aldrich) supplemented with 10% FBS, 50 μg ml−1 streptomycin, 50 units ml−1 penicillin and 50 U ml−1 IL-2.
Lentivirus production
HEK 293T cells (0.8 million) were seeded in a 6-well plate (Day 1) and incubated overnight. Cells in each well were co-transfected (Day 2) using X-tremeGENE HP (Roche) with 0.8 μg of the appropriate lentiviral transfer plasmid encoding an antigen receptor (1G4 TCR or c259 TCR) and the lentiviral packaging plasmids: pRSV-Rev (0.25 μg), pMDLg/pRRE (0.53 μg) and pVSV-G (0.35 μg). The media were replaced 18 h following transfection (Day 3). At 24 h after the media exchange, the supernatant from one well was collected, filtered and used for the transduction of 1 million human T cells (Day 4).
Production of TCR transduced primary human T cells
T cells were isolated from anonymized leucocyte cones (Day 3) purchased from the NHS Blood Donor Centre at the John Radcliffe Hospital (Oxford University Hospitals). Due to the anonymized nature of the cones, biological sex and gender were not variables in the present study and were therefore randomized, hence the authors were blinded to these variables. RosetteSep Human CD8+ Enrichment Cocktail (STEMCELL Technologies) was used for cytotoxic T cells, or CD4+ T Cell Enrichment Cocktail (STEMCELL Technologies) for helper T cells. The enrichment cocktail was added at 150 μl ml−1 of sample and incubated at r.t. for 20 min. The sample was diluted with an equal volume of PBS and layered on Ficoll Paque Plus (Cytiva) density gradient medium at a 0.8:1 ratio (Ficoll:sample).
The sample was centrifuged at 1,200 g for 30 min (brake off). Cells at the interface of the Ficoll media and plasma were collected (buffy coat) and washed twice (centrifuged at 500 g for 5 min). Cells were resuspended in complete RPMI media supplemented with IL-2 (50 U ml−1) at a density of 1 million cells per ml. Dynabeads Human T-Activator CD3/CD28 (ThermoFisher) were added (1 million beads per ml) and cells were incubated overnight.
One million cells were transduced with the filtered lentiviral supernatant (Day 4). On Day 6 and on Day 8, 1 ml of medium was removed and replaced with 1 ml of fresh medium. On Day 9, Dynabeads were removed using a magnetic stand (6 days following isolation). Cells were resuspended in fresh media every other day at a density of 1 million per ml and used for co-culture experiments. At 17 days following isolation, T cells were discarded.
CRISPR/Cas9 knockout of T cell proteins
Cas9 ribonucleoproteins (RNPs) were prepared by mixing 8.5 μg of TruCut Cas9 protein v2 (ThermoFisher) with 150 pmol of sgRNA mix (Truguide Synthetic gRNA, ThermoFisher) for each target gene (Supplementary Table 4) and Opti-MEM (Gibco) to a final volume of 5 μl. The RNPs were incubated for 15 min at r.t.
One million freshly isolated T cells were washed with Opti-MEM (Gibco) and resuspended at a density of 20 million per ml. The T cells were mixed with the RNPs and transferred into a BTX Cuvette Plus electroporation cuvette (2 mm gap, Harvard Bioscience). The cells were electroporated using a BTX ECM 830 Square Wave Electroporation System (Harvard Bioscience) at 300 V for 2 ms. Immediately following electroporation, the cells were transferred to complete RPMI media supplemented with IL-2, and Dynabeads Human T-Activator CD3/CD28 (ThermoFisher) were added.
Negative selection of T cell knockout cells
T cells with residual target protein expression were depleted by antibody staining and bead pulldown. T cells were resuspended in MACS buffer (PBS, 0.5% BSA, 2 mM EDTA) at a density of 100 million cells per ml. Cells were stained with 5 μl of the corresponding PE-labelled antibody per million cells for 15 min at 4 ∘C, washed with MACS buffer and resuspended at a density of 100 million cells per ml. A volume of 1 μl of MojoSort anti-PE nanobeads (Biolegend) was added per million cells and incubated on ice for 15 min. The cells were washed with MACS buffer and the beads were pulled down magnetically. The supernatant containing the negatively selected cells was collected.
Cellular co-culture assays
U87 cells (50,000) in 100 μl of DMEM were seeded per well in a 96-well flat-bottom plate and incubated overnight. Alternatively, 100,000 T2 cells were placed in each well of a 96-well flat-bottom plate. Peptides were diluted in DMEM to the appropriate concentration, added to each well containing cells and incubated for 60 min at 37 ∘C and 10% CO2. The media were discarded and 50,000 T cells were added to each well in 200 μl of RPMI medium. Cells were incubated for 20 h at 37 ∘C and 5% CO2. Supernatants were collected for cytotoxicity and ELISA analysis. A volume of 25 μl of 100 mM EDTA PBS was added to each well containing the cells and samples were incubated for 5 min at 37 ∘C and 5% CO2. Cells were detached by thoroughly pipetting each well and transferred to a 96-well V-bottom plate.
Lck chemical inhibition assay
U87 cells (50,000) in 100 μl of DMEM were seeded per well in a 96-well flat-bottom plate and incubated overnight. T cells were treated with the appropriate concentration of A-770041 for 1 h. The DMEM media were discarded and 50,000 A-770041-treated T cells were added to each well in 200 μl of RPMI media. Cells were incubated for 4 h at 37 ∘C and 5% CO2. Supernatants were collected for cytotoxicity and ELISA analysis, and T cells were analysed for activation markers as in other co-culture assays.
Flow cytometry
The following fluorophore-conjugated mAbs were used: CD45 (Biolegend, clone HI30), CD3 (Biolegend, clone OKT3), 4-1BB (Biolegend, clone 4B4-1), CD69 (Biolegend, clone FN50), CD8α (Biolegend, clone HIT8), CD4 (Biolegend, clone RPA-T4), CD43 (Biolegend, clone CD43-10G7), CD11α (Biolegend, clone TS2/4), CD5 (Biolegend, clone UCHT2), CD2 (Biolegend, clone TS1/8) and TCR Vβ13.1 (Biolegend, clone H131).
Cells were stained for 20 min at 4 ∘C, washed with PBS and analysed using a BD X-20 or a Cytoflex LX flow cytometer (Beckman Couter). Data were analysed using FlowJo v.10, RRID:SCR008520 (BD Biosciences) and GraphPad Prism, RRID:SCR002798 (GraphPad Software).
Cytotoxicity assay
Target cell lines were engineered to express the Nluc luciferase57. A 2 mM coelenterazine (CTZ) stock solution was prepared in methanol, aliquoted and stored at −80 ∘C. Supernatant from co-culture assays was mixed at a 1:1 ratio with PBS 10 μM CTZ, and luminescence was read using a SpectraMax M3 microplate reader (Molecular Devices).
Cytokine ELISA
Invitrogen Human IFNγ ELISA kits (ThermoFisher) were used following manufacturer protocol to quantify levels of cytokine in diluted T cell supernatant. A SpectraMax M3 microplate reader (Molecular Devices) was used to measure absorbances at 450 nm and 570 nm.
Longitudinal killing assay
mCherry positive A375 cells were seeded in a 96-well flat-bottom plate and incubated overnight in 100 μl of DMEM at 37 ∘C and 5% CO2. To normalize differences in TCR transduction across different batches, 50,000 a3a or c259 TCR positive T cells were used as the starting concentration, and they were serially diluted to the appropriate effector:target (E:T) ratios. For each E:T ratio, 100 μl of T cells were plated in triplicate. The mCherry positive A375 cell number was quantified every 2 h using an xCELLigence RTCA eSight system (Agilent).
Surface plasmon resonance
All SPR experiments were carried out at the Dunn School SPR facility using our published method24. The c259 TCR/pMHC steady-state binding affinities were measured on a Biacore T200 SPR system (GE Healthcare) with a CAP chip using HBS-EP as running buffer. The CAP chip was saturated with streptavidin and biotinylated pMHCs were immobilized to the desired level. A titration of the TCR was flowed through at 37 ∘C. The reference flow cell contained CD58 immobilized at levels matching those of pMHCs on the remaining flow cells. The signal from the reference flow cell was subtracted (single referencing) and the average signal from the closest buffer injection was subtracted (double referencing). Steady-state binding affinity was calculated by fitting the one site-specific binding model (Response = Bmax [TCR]/(KD + [TCR])) on GraphPad Prism to double-referenced equilibrium resonance units (RU) values. The Bmax was constrained to the inferred Bmax from the empirical standard curve generated by plotting the maximal binding of a conformationally sensitive pMHC antibody to the maximal TCR binding (Bmax).
Pooled 9-mer peptide library
A library of pooled randomly synthetized 9-mer peptides was produced by Peptide Protein Research. This library was composed of all natural amino acids, except cysteine, as previously described58. The library has a theoretical diversity of 199 peptides.
U87 cells (50,000) in 100 μl of DMEM were seeded per well in a 96-well flat-bottom plate and incubated overnight. The 9-mer pooled peptide library was diluted in DMEM to 100 μM, added to each well containing cells and incubated for 60 min at 37 ∘C and 10% CO2. T cells (50,000) were added to each well in 200 μl of RPMI medium. Cells were incubated for 20 h at 37 ∘C and 5% CO2. Supernatants were collected for cytotoxicity analysis. In each independent biological experiment, three technical measurements were taken and averaged.
Positional scanning peptide library SPR
To prepare pMHC complexes presenting the local peptide library, a disulfide-stabilized variant of the human MHC-I protein HLA-A*02:01 (DS-A2) was used59. The DS-A2 protein was produced as described previously59. Briefly, the DS-A2 and β2-microglobulin (β2m) subunits were produced in E. coli as inclusion bodies and solubilized in 8 M urea. The protein was then refolded in the presence of GlyLeu, a dipeptide that binds with low affinity to the peptide-binding cleft. The refolded DS-A2–β2m complexes were purified by size exclusion chromatography on a Superdex S75 10/300 column (GE Healthcare/Cytiva) in HBS-EP buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.05% Tween 20). Local-library peptides were loaded by incubating the DS-A2–β2m complex with each peptide for 2 h at r.t. The pMHC complexes were stored at 4 ∘C until use within 24 h.
Soluble c259 TCR was produced as separate TCRα and TCRβ chains in E. coli. Both chains were recovered as inclusion bodies, solubilized in 100 mM Tris-HCl (pH 8.0), 8 M urea and 2 mM dithiothreitol, and then stored in aliquots at −70 ∘C. For refolding, 30 mg of each TCR chain was added to 1 l of refolding buffer (150 mM Tris-HCl (pH 8.0) 3 M urea, 200 mM Arg-HCl, 0.5 mM EDTA, 0.1 mM PMSF) and stirred for 1 h at 4 ∘C. This was followed by dialysis in 10 l 10 mM Tris-HCl (pH 8.5) buffer for 3 days in total, with the dialysis buffer changed after 1 day. The refolded c259 TCR was purified using anion exchange chromatography (HiTrap Q HP, Cytiva), followed by size exclusion chromatography (Superdex 200 Increase, Cytiva) in HBS-EP buffer. Purified c259 was used within 48 h.
High-throughput affinity measurements of c259 TCR binding to MHC loaded with the peptide library were performed using LSA or LSAXT (Carterra). Each pMHC was immobilized via biotin–streptavidin binding on a different spot of the SAHC30M biosensor (Carterra) for 20 min, resulting in immobilization levels between 200 and 900 RUs. Measurements were performed in HBS-EP buffer at 37 ∘C. A 2-fold dilution series of c259 TCR was prepared in HBS-EP buffer, with the highest concentration between 100–130 μM. Starting with the highest dilution, increasing concentrations of c259 were injected over the chip for 5 min, followed by 5–10 min of dissociation, without regeneration. Afterwards, a β2m specific antibody (clone B2M-01 (ThermoFisher) or BBM.1 (Absolute Antibody)) was injected for 10 min. The resulting data were analysed using Kinetics Software (Carterra). Any spikes were removed from the data before referencing against empty control spots or spots immobilized with CD86 at matching immobilization levels. The final injection in a series 6 buffer injections before TCR injection was subtracted from the data for double referencing. Subsequently, the steady-state binding RU was calculated by taking the average RU from over 20 s.
Steady-state analysis was performed to obtain the TCR–pMHC affinity (KD) values. First, steady-state data were fitted with a one site-specific binding model (Response = Bmax [TCR]/(KD + [TCR])), with KD and Bmax unconstrained. We then constructed an empirical standard curve using high-affinity pMHCs (KD < 20 μM) to relate maximal anti-β2m binding to TCR Bmax. Next, steady-state data for all pMHCs were fitted with a one site-specific binding model, with Bmax constrained to the Bmax inferred from the empirical standard curve. We excluded KD values for peptides, where we observed little or no anti-β2m binding responses, indicating that the pMHC complex was unstable and lost the peptide over time (indicated as N/A in Supplementary Table 2). We further excluded KD values for pMHC that produced a TCR binding response of less than 5 RU (indicated as non-binders (NB) in Supplementary Table 2).
Data analysis
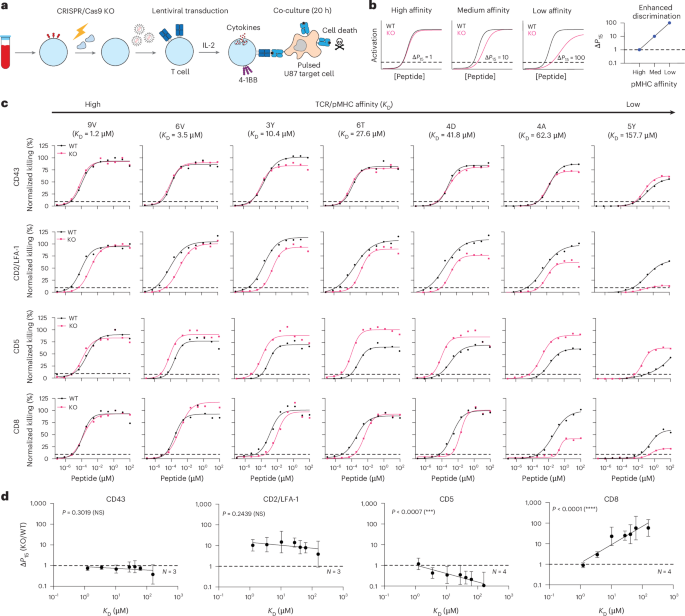
EC50 was calculated as the concentration of antigen required to elicit 50% of the maximum response determined for each condition individually, whereas P15 was calculated as the concentration of antigen required to elicit 15% of the maximum activation for the experiment.
We have used P15 for two reasons. First, P15 always corresponds to the concentration of peptide required to activate 15% of T cells, independent of the maximum responses. In contrast, EC50 is the concentration of peptide required to activate 50% of the maximum response (that is, normalized to the maximum of wild type or knockout). In other words, two antigens with the same antigen potency as measured by EC50 values may produce a different percentage of activated T cells if their maximum response (Emax) differ. In this case, the antigens would have different antigen potencies as defined by P15. Second, the use of P15 does not require the dose–response curve to saturate, enabling accurate estimates of P15 from lower-affinity interactions. This measure of potency was previously used in ref. 24 to study ligand discrimination.
The study is largely focused on comparing antigen sensitivity using EC50 or P15 measures, which we have found to display standard deviations of 0.2 (on log-transformed values). The smallest effective size that we aimed to resolve was 3-fold changes (a difference of 0.47 on log-transformed values), and a power calculation shows that this can be resolved with a power of 80% (⍺ at 0.05) using three samples in each group. Therefore, all experiments relied on a minimum of 3 independent donors.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.