Study setting
A hospital-based prospective cohort study was carried out with support from the WHO at five university hospitals affiliated with Al-Azhar University, Egypt, focusing on HWs. Three of these hospitals are in the Cairo governorate (representing the capital), one in the Damietta governorate (representing Lower Egypt), and one in Assiut governorate (representing Upper Egypt). The WHO’s protocol, design, and methodology for assessing COVID-19 vaccine effectiveness were customized to suit the specific context and settings of the country.
Study participants
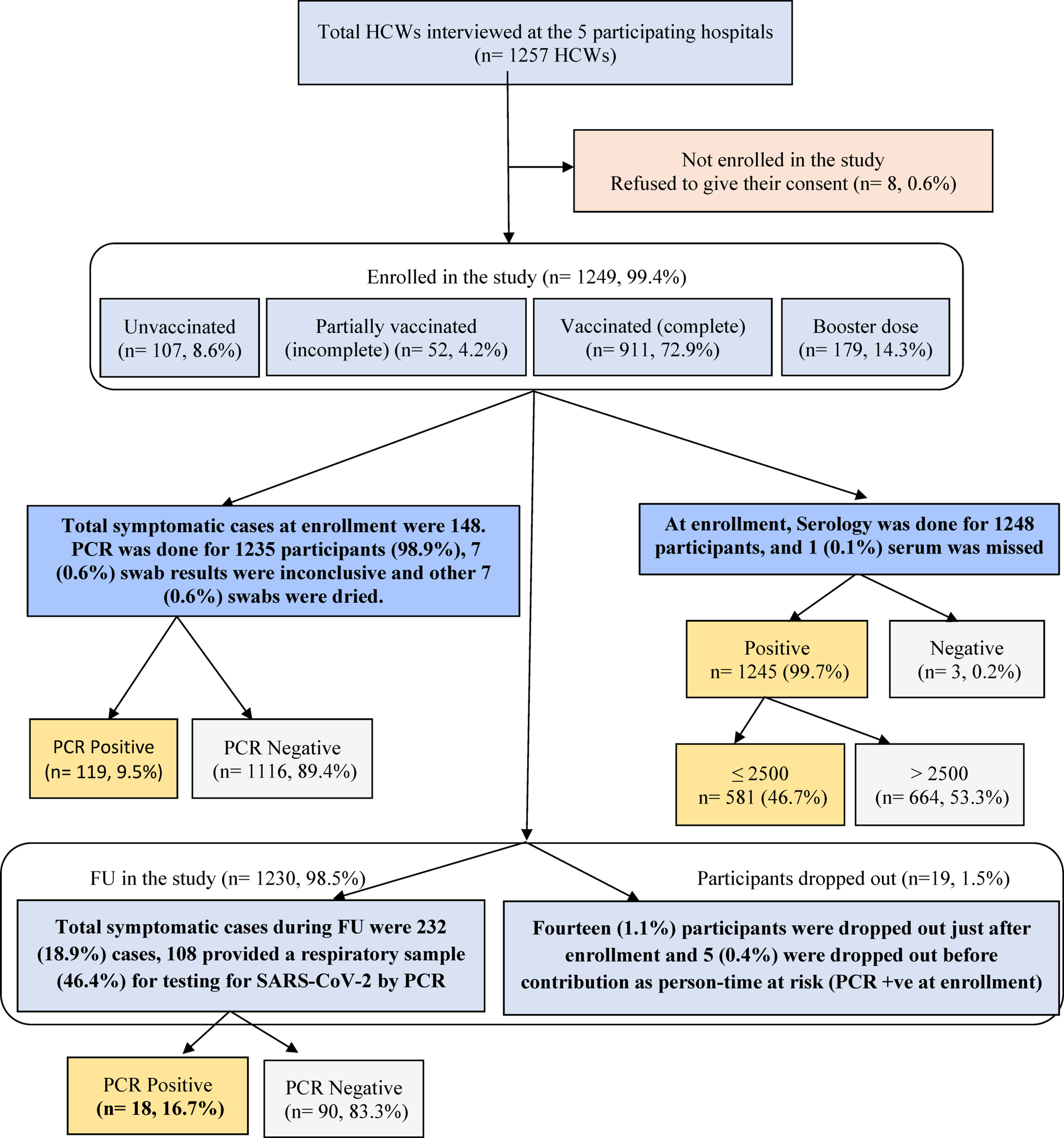
HWs affiliated with Al-Azhar University Hospitals were randomly selected, regardless of their COVID-19 vaccination status. All categories of HWs at these hospitals were eligible for inclusion in the study, provided informed consent was obtained. Eligible participants include those who interact with patients, handle their body specimens, or manage potentially infectious waste. This encompassed physicians, nurses, emergency medical personnel, laboratory technicians, and administrative staff. To participate, HWs had to be eligible for vaccination and have no contraindications to receiving the COVID-19 vaccine. Health worker who had already received the COVID-19 vaccine through the national vaccination program outside their hospitals were also included, provided that comprehensive vaccination details were available. Those who declined to provide informed consent or were ineligible for COVID-19 vaccination due to conditions such as severe allergies or pregnancy were excluded from the study. The number of participants from each facility was estimated according to the total workforce size at each hospital: 300 participants from each hospital in Cairo, 200 from the hospital in Damietta, and 150 from the hospital in Assiut, with the total work force being 13,260 HWs in the five hospitals.
Sampling criteria
The sampling frame for the study was derived from the updated healthcare workforce data at each university hospital, and to ensure adequate representation of specific subgroups within the workforce, stratified random sampling was conducted at each hospital. The sampling process was guided by probability proportional to size, based on job roles. A sampling interval was then calculated, and a random starting point was chosen using a table of random numbers. Out of this list, the participating HWs in each subgroup were selected based on the subgroup’s population size within each hospital.
Participant enrollment and follow-up procedures
The study objectives were explained to HWs at the participating hospitals. Participation was voluntary, and participants could withdraw at any time without penalties, though they were asked to notify the team if they chose to do so. Enrollment took place from July to August 2022.
A face-to-face interview was carried out with each participant and a questionnaire was administered after signing an informed consent. This questionnaire included demographic, medical history, COVID- 19 vaccination history, previous COVID- 19 infections, symptoms developed and way of its diagnosis within the past year and 14 days before the interview as well as occupational and community-related behaviors. Regular ZOOM meetings were held with the research team to ensure proper study implementation at each hospital and to monitor adherence to the study protocol by the team. Participants’ self-reported vaccination status was verified through sources such as occupational health records, vaccination cards, or vaccine registries. The Ministry of Health and Population (MOH&P) provided each vaccine recipient with a card detailing their vaccine information, and vaccines were distributed to the university hospitals through a cold chain system. Participants were registered online [http://www.egcovac.mohp.gov.eg], and vaccine was free of charge. Each participant who consented to take the vaccine had to fill out an application form and vaccination was postponed for 3 months for those who had a recent COVID-19 infection. Participation was encouraged through a national advertising campaign.
Follow-up phase
Participants in each hospital were grouped (20-25) for each investigator, who had a list of his / her assigned participants’ contact details. Participants were monitored biweekly by investigators to complete follow-up questionnaires. Regular reminders were sent before follow-up appointments. The objectives of the follow-up were to identify newly developed COVID − 19 cases among the participating HWs, track changes in their vaccination status, and to monitor changes in their potential exposure risks. Those who failed to respond within 48 h of their follow-up appointments were contacted again via an alternative way of communication. If they declined to continue, their reasons were documented. During follow up, COVID- 19 infection was identified if any participant met the following WHO COVID-19 case definition; acute onset of fever and cough OR acute onset of any three or more of the following symptoms in the previous 7 days: fever, cough, general weakness/fatigue, headache, myalgia, sore throat, coryza, dyspnea, anorexia/nausea/vomiting, diarrhea, altered mental status, anosmia, or ageusia [19]. In case of COVID-19 symptoms, the investigator examined the participant and nasopharyngeal swabs were taken within 24–48 h following the appearance of symptoms. Breakthrough infection is the term used to describe developed infections in fully vaccinated people, while non-breakthrough infection refers to infections in unvaccinated people [20] All follow-up interactions were documented using standardized written records to ensure consistency in data collection.
Data collection procedures
The WHO provided financial and technical support for methodology, data management, report development and statistical analysis of the data [21, 22]. The data collection forms were based on the WHO protocol and relevant questionnaires [23]. Five questionnaire forms were included, each customized to fit the study requirements. The pre-enrollment and enrollment questionnaires were filled at enrollment and the follow-up questionnaire during the follow-up period. The laboratory questionnaires (serology/virology) included date, type of specimens, assigned test and results.
Sample size calculation
The sample size was calculated based on Table 2 in the WHO protocol [24] Assuming a vaccine effectiveness of 60% and that 20% of unvaccinated HWs would be infected with SARS-CoV-2 over a period of 12 months and vaccination coverage among HWs of 90%, the required minimum sample size of 1006 participants was calculated. Accounting for an expected 20% loss to follow-up during the one-year follow-up period, 1250 participants ultimately needed to be included.
Ethical considerations
Each participant was allocated a unique study ID number with a barcode at enrollment which was scanned at all subsequent steps to identify each respective individual’s documents and test. Name and national ID number were included in study databases. Personal identifying information was maintained only by the person responsible in each study site in accordance with regulatory agencies requirements. To ensure confidentiality, anonymization techniques were implemented by removing sensitive data including personally identifiable information, implementing safeguards against participant identification during data entry. The study was cleared by the Ethical Committee of Al- Azhar University on 5/12/2021 (AU-REC-2021-0002).
Laboratory investigations
Nasopharyngeal swabs were transported to the main Virology Laboratory- Medical Microbiology Department, Faculty of Medicine(for girls)-Al-Azhar University and stored at −80 °C till evaluation for the presence of SARS-CoV-2 RNA via real-time RT‒PCR, whereas serum samples were transported to Clinical Pathology laboratories and stored at −20 °C, at Al-Zahraa, Al-Hussein and Bab-AlSharia Hospitals in Cairo for SARS-CoV-2-binding antibody quantitation. Follow-up NP samples were sent directly to the main virology laboratory or stored at −20 °C for a few days and then transferred to the laboratory in Cairo for analysis.
Detection of SARS-CoV-2 binding antibodies
Total antibodies against the receptor binding domain (RBD) of the S protein were quantified in serum samples collected at enrollment via the Roche Elecsys Anti-SARS-CoV-2 S immunoassay on a Roche Cobas e 411 (Roche Diagnostics, GmbH, Germany). The limits of the blank (LoB) and limit of detection (LoD) were 0.30 U/mL and 0.40 U/mL, respectively. Test results < 0.8 U/mL were classified as nonreactive, whereas those ≥ 0.8 U/mL were classified as reactive. The upper limit of the kit was 250 U/ml, and due to a high number of positive samples that were out of range, all samples were diluted 1:10 to obtain values ≤ 2500 U/ml.
Real-time RT‒PCR
Reverse-transcriptase polymerase chain reaction (RT-PCR) testing for SARS-CoV-2 was conducted on NP samples. This was carried out using a qualitative real-time RT-PCR assay kit (the artus® SARS-CoV-2 Prep and Amp UM Kit -QIAGEN-Germany) targeting 2 viral genes (N1 and N2 of the N gene were detected through the same fluorescence channel). The two targets were not differentiated, and amplification of either or both targets led to the generation of fluorescence signal genes. The sample preparation and detection steps were integrated into a single kit with a limit of detection of 950 cp/ml. A sampling control (RNase P) and internal RNA control, together with positive and negative external controls, were included. Cycling conditions was adjusted according to the following:10 min at 50 °C for R-T reaction, followed by 2 min at 95 °C for initial activation and 40 cycles of five seconds at 95 °C and 30 s at 58 °C. Positive samples were considered when the CT was above 38 and samples were considered invalid if the channel for housekeeping gene was negative.Invalid results were obtained for a noticeable number of samples. Thus, RNA extraction (QIAamp DSP Virus spin kit) was performed for theses samples, where a larger sample volume was tested (200 µl instead of 10 µl), adequate extraction and purification of viral RNA by enzymatic lysis and mixing (protease + buffer AL with carrier RNA), and then RT‒PCR steps with the artus® SARS-CoV-2 Prep and Amp UM Kit were conducted (bypassing its extraction step). The results were conclusive.
Statistical analysis
Data entry into the REDCap platform occurred under the guidance and support of the WHO-EMRO technical team. Participant’s name, address, phone, and mobile number were excluded, as this information was kept only with the follow-up team.
Statistical analysis was carried out using SPSS for Windows, version 23. Categorical variables were presented as numbers and percentages, and continuous variables as medians and IQRs. Different statistical methods were used to assess the significance level for the differences between the study groups according to their vaccination status. Chi-square was used to detect the significant difference between categorical variables, and Fisher’s exact test was used if the expected number was below 5 in any cell. For continuous variables, one-way ANOVA was used for parametric data and the Kruskal-Wallis test for non-parametric data. Survival analysis was carried out using the Kaplan-Meier test, and a curve was generated to inspect the cumulative incidence according to vaccination status.
The incidence of SARS-CoV-2 infection was calculated as the number of events reported during the person-time at risk. For each participant, contribution of person-time at risk started from the time of enrolment, 90 days after having a SARS-CoV-2 infection confirmed by RT-PCR at or before enrolment, or 14 days after receiving 1 or 2 doses of the vaccine or 7 days from receiving booster dose, whichever was the latest. We considered 4 weeks until being at risk, for participants with a previous infection detect by only positive serology at enrolment. Person-time at risk ended on the date of onset of COVID-19 symptoms confirmed by positive RT-PCR test, the date of lost follow-up, or the date of end of the study on 15-8-2023.
Exposure status classification and grouping for vaccine effectiveness analysis
Exposure status was classified based on the participants’ COVID-19 vaccination history at the time of being at risk. There was no change in vaccination status from enrolment to the end of follow up except for only one participant, who had received his third dose in September 2022. Participant was considered vaccinated with the first dose, 14 days after receiving the first vaccine dose (partial vaccination for all studied vaccines except mRNA-1273(Moderna) vaccine and fully vaccinated 14 days after receiving the second dose of the vaccine (for all studied vaccines except one dose only for mRNA-1273(Moderna) vaccine, and for booster after 7 days after receiving the third dose of vaccine.
Symptomatic RT-PCR confirmed SARS-CoV-2 infection was the primary outcome. Vaccine effectiveness (VE) was estimated using the unvaccinated as a reference group, unless stated otherwise. Participants were grouped according to their vaccination status, type of vaccine and time since vaccination. VE% was estimated using Cox regression proportional hazards models; hazard ratios comparing vaccinated and unvaccinated were estimated with vaccination as a time-varying exposure. VE% = 1– hazard ratio [HR]* 100, where the HR = Exp (B) in the Cox regression model. Follow-up time was from baseline to the time of symptomatic SARS-CoV-2 confirmed with positive RT-PCR or study exit. The 95% CI of VE was computed based on the 95% CI of the HR in the Cox regression.
Subgroup analyses for VE by vaccination doses (partial, fully vaccinated, and booster dose) vs. unvaccinated, different vaccine type vs. unvaccinated, then fully vaccinated vs. unvaccinated and vaccine type regarding the prior infection) were performed. For VE calculation, prior infection depends on RT-PCR positivity, either before or at enrollment, or positive serology at enrollment for unvaccinated participants. However, for prior infection, 15 participants were excluded when calculating the VEs, as 14 of them had missing RT-PCR results at enrollment (7 sample results were inconclusive / invalid and 7 were dried samples) and one missing a serology sample. These missing RT-PCR results were at random, from different hospitals.
An additional analysis was planned to stratify VE% estimates by time since vaccination. Follow-up was identified from the start of being at risk to the earliest of outcome or study exit. In the current study, the median duration from receiving the 2nd dose till the end of follow-up was 631 days (IQR: 557–730 days), and between the booster dose and the end of follow-up was 394 days (IQR: 319–491 days). Thus, we only stratified the duration into ≤ 365 days and > 365 days.
Confounding factors and effect modifiers
Both unadjusted and adjusted estimates of VE were presented. Adjustment was made in the Cox regression model for potential confounders (age, sex, chronic comorbidities, and health facility). Hospitals were categorized in two: Cairo hospitals (Al-Zahraa, Al-Hussein, and Bab-Alsharia) and peripheral (Damiatta and Assiut). Adjustment was made in the multivariable cox regression model for all potential confounders. None of these variables were found to be significantly changing the VE% after adjustment, when multivariate Cox regression backwards Wald analysis was carried out.
Sensitivity analysis
Sensitivity analyses were performed to estimate the robustness of the VE estimates produced in the main analysis against different assumptions (e.g., assumptions made on missing data) and/or sources of bias (e.g., the effect of unmeasured confounding factors).
Based on the assumptions made and on the different sources of bias identified, different sensitivity analyses were performed:.
● Excluding participants with shorter than expected vaccination intervals between the 1st and 2nd dose and including those with < 21 days between the first and second dose of vaccine.
● Consider participants were at risk after 60 days instead of 90 days after the previous infection that detected by RT-PCR.
● Exclusion of symptomatic patients who refused to give nasopharyngeal swabs versus including them in the VE% estimation.