Plant material and PVY strain
The PVY isolate used in this study was obtained from infected potato plants (Isolate MN21, strain PVYN−Wi, cordially provided by Dr. Stewart Gray from USDA-ARS/Cornell University). The isolate was maintained in lyophilized tobacco plant tissue and stored at -80 ˚C. For inoculum reactivation, tobacco plants (Nicotiana tabacum L. cv. Samsun, kindly provided by the USDA National Plant Germplasm System – USA074), propagated via seed, were kept in 1-gallon pots containing Cornell potting mix growth [22] and maintained in an insect-free growth chamber under controlled conditions at 25 ± 3oC and 16 h photoperiod. When the plants were at the three- to five-leaf stage, they were mechanically inoculated with PVY using carborundum (325 mesh) as an abrasive. The inoculum was prepared from lyophilized tissue homogenized in phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4, with pH adjusted to 7.4 with HCl) at 1:5 w/v (1 g of leaf to 5 ml of PBS). Seven days after inoculation, characteristic symptoms of PVY infection could be observed on inoculated plants, and the PVY strain identity was confirmed using a multiplex RT-PCR [23]. Infected plants were maintained in a growth chamber, per the conditions described above, and used as viral inoculum sources in subsequent experiments.
Small RNA profiling analyses
Small RNA profiles were analyzed to aid the selection of PVY genomic regions for testing, as performed by our group previously [13]. Infected tobacco leaves from source plants were harvested, ground in PBS at 1:5 w/v (1 g of leaf to 5 ml of PBS), and used as PVY inoculum. Then, three tobacco plants (at the three- to five-leaf stage) were inoculated by using a cotton swab to lightly rub the PVY inoculum using carborundum as an abrasive. The top two fully mature leaves were treated with PVY inoculum and PBS alone as a mock, on individual plants as two different treatments, each treatment had three replicates. One-week post-inoculation, a systemically infected leaf was harvested from each individual plant and immediately frozen in liquid nitrogen, followed by total RNA extraction using the Purelink Plant RNA reagent, following the manufacturer’s instructions (Invitrogen, Waltham-MA, USA).
Small interfering RNAs (siRNAs) were isolated from the total RNA using polyacrylamide gel electrophoresis, followed by adapter ligation to the 5′ and 3′ ends of the siRNAs. The NEBNext Small RNA Library Prep Set for Illumina (NEB, Ipswich – MA, USA) was then used according to the manufacturer’s protocol. Libraries were barcoded, pooled in equal molarity, quality checked using a 2100 Bioanalyzer, and then sequenced on an Illumina HiSeq4000 platform (generating 76 bp single-end reads) at Yale Center for Genome Analysis, Yale University, New Haven – CT, USA. Then, Illumina’s CASAVA pipeline v1.8.2 software was used to demultiplex the raw reads, followed by read quality enhancement using Trimmomatic software [24]. Identification of distinctive virus-derived small interfering RNA (vsiRNA) reads was performed by aligning the reads with the PVY genome with a specific pipeline [25]. VsiRNA orientation and graphical representation across the viral genome were performed using MISIS [26] and R software [27]. An in-house script was created to select the regions of high abundance of vsiRNAs to design the dsRNAs for this study (S1 Table). The script was fed with vsiRNA data described above and given the genomic locations to scan (PVY cistrons HC-Pro, NIb, and CP) and the size of the dsRNA to be designed (∼ 600 bp).
DsRNA synthesis
Total RNA extracted from an infected tobacco plant was reverse transcribed using the ProtoScript II First Strand cDNA Synthesis Kit (NEB, Ipswich, MA), following the manufacturer’s instructions. The cDNA obtained was amplified using PVY-specific primers designed for the CP, HC-Pro, and NIb cistrons (Table 1), using the Phusion High-Fidelity PCR Master Mix with HF Buffer (NEB, Ipswich, MA). These genomic regions were selected based on the siRNA mapping analyses data, we selected regions with a high concentration of 21-nt vsiRNAs. Amplicons generated (∼ 600 bp) for each genomic region were resolved in 1.5% agarose gel electrophoresis, cleaned using ChargeSwitch PCR Clean-Up Kit (Thermo Fisher Scientific, Waltham, MA), quantified using Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA), and Sanger sequenced at the Keck Biotechnology Resource Laboratory, Yale University, New Haven – CT, USA, to confirm the amplicon sequences prior to dsRNA synthesis. Then, PCR amplicons were used as templates for dsRNA synthesis using the T7 Ribomax Express RNAi System kit (Promega, Madison, WI), following the manufacturer’s instructions. The T7 RNA polymerase promoter (TAATACGACTCACTATAGGG) was added at the 5’-end of the amplification primers to increase the effectiveness of the RNA synthesis.
Viral inoculum standardization
To determine the minimum inoculum concentration required to induce PVY infection symptoms in tobacco plants, a sequential inoculum dilution experiment was conducted. Infected tobacco leaves were collected and macerated at 1:1 w/v (1 gram of leaf to 1 ml of PBS) in an extraction bag (Bioreba, Reinach BL, Switzerland), which possesses a synthetic intermediate layer for optimal filtration, to create an undiluted stock solution. Then, serial dilutions (1:100, 1:200, 1:400, 1:800, 1:1200, 1:1600, and 1:2000) from the stock were made by adding PBS. A double antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA) was performed on the diluted samples to determine the absorbance value for each diluted point, ELISA was duplicated following the manufacturer’s instructions (Bioreba, Reinach BL, Switzerland). Then, a bioassay was set up where five tobacco plants (at the three- to five-leaf stage) were inoculated per dilution point. The top two fully mature leaves were inoculated by using a cotton swab to lightly rub the PVY inoculum, 50 µl of inoculum per leaf, using carborundum as an abrasive, five plants were inoculated with PBS only as the mock. PVY symptoms in the plants were evaluated at seven- and 14-days post-inoculation and correlated to the absorbance values for each dilution. ELISA was performed on inoculated plants at every evaluation time to determine the plant infection status.
Effect of synthesized DsRNAs on suppressing PVY infection
To test the effect of synthesized dsRNA from each cistron in suppressing PVY infection in tobacco plants, 50 µg of dsRNAs were applied per leaf, and two leaves were treated per plant. Plants were mechanically inoculated with PVY either along with the dsRNA (1 day) or at five days after dsRNA treatment. The experiment included two controls, a positive control consisting of plants inoculated with PVY only (no dsRNA), and a negative control (Mock) where plants were treated with dsRNA only (no PVY) – we did set up a pilot experiment using dsRNA from the Fusarium oxysporum FpPPR1 gene to verify if a non-PVY-target dsRNA would induce some level of PVY suppression, and since we detect no effect of applying such dsRNA on PVY suppression, we did not include this control in our experiments. Plants were maintained in an insect-free growth chamber at 25 ± 3 ◦C and a 16 h photoperiod. Two weeks after viral inoculation, PVY symptoms were assessed, and ELISA was conducted to check the infection status of each plant (Fig. 1). This experiment was repeated three times with five replicates per treatment.
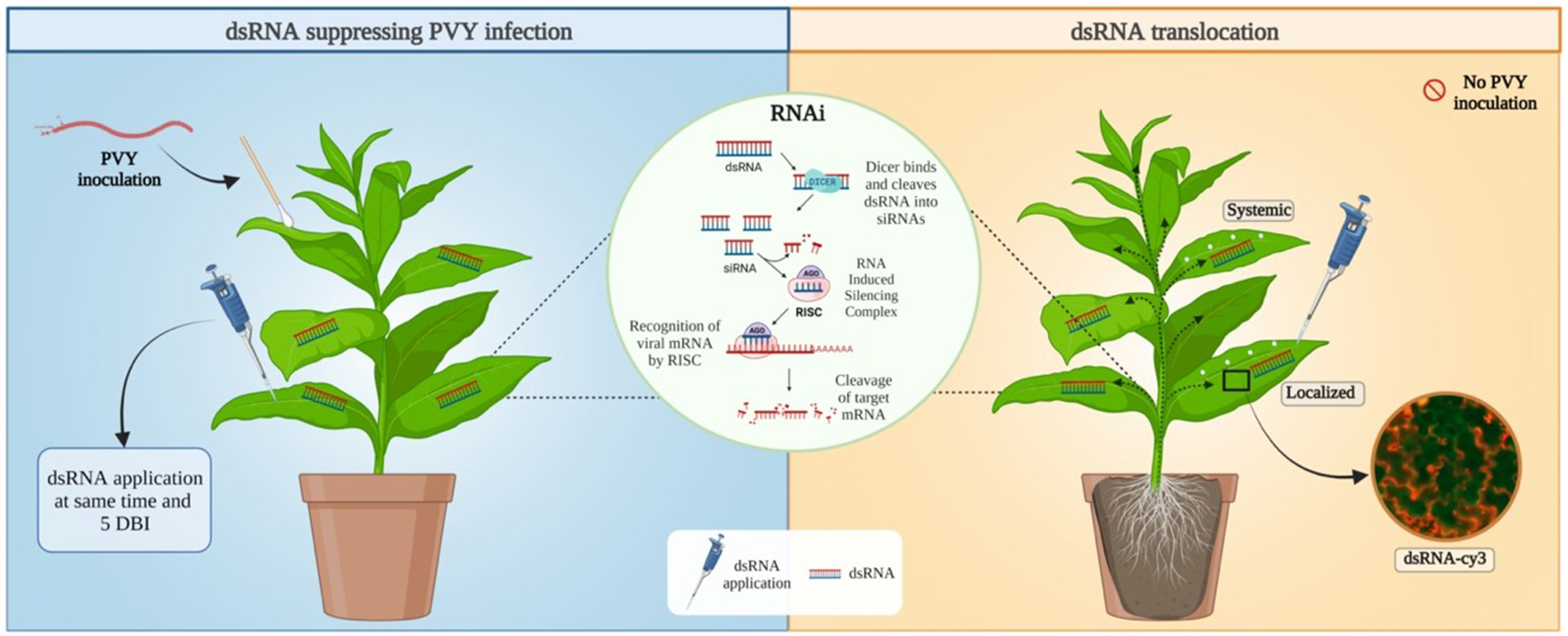
Schematic illustration summarizing the mechanism and the fate of foliar exogenous dsRNA application in a tobacco plant. The left Section depicts the suppression of PVY infection using dsRNA. The right section depicts the movement of the dsRNA inside the tobacco plant. The middle circle depicts the molecular mechanism of RNA interference (RNAi). DBI = Days before infection with potato virus Y (PVY). Created with BioRender.com
DsRNA translocation in tobacco plants
To shed light on how exogenously applied dsRNA molecules behave inside the plant and their potential effects in suppressing PVY infection via RNAi, the translocation of the applied dsRNA molecules inside the plants was evaluated. The dsRNAs synthesized from CP, HC-Pro, and NIb cistrons were applied as previously mentioned (50 µg of dsRNA were applied per leaf, and two leaves were treated per plant), with the exception that no virus inoculation was performed (S1 Fig, A and B). The mock treatment used was only nuclease-free water without dsRNA; there were three plants per treatment. Since the symplastic movement of mobile RNA in plants occurs from the photosynthetic source tissue (old leaves) to photosynthetic sink tissue (young leaves) [28], dsRNA was applied to the two fully formed leaves of plants at the three- to five-leaf stage. Three-millimeter discs were collected every 24 h (S1 Fig C) from the dsRNA-treated leaves (localized leaf) and the subsequent young leaf near the application site (systemic leaf). This process was repeated on the same leaves for 14 days. Immediately after collection, the discs were washed in a 0.05% tween 20 solution to remove potential dsRNAs present on the outside of the discs and dried on a paper towel. In a pilot study, we performed RT-PCR on the solution from the washes with 0.05% tween 20, and no dsRNA residue could be detected in the solution from the second, third, and fourth washes; dsRNA residual was detected in the solution from the first wash. To play safe, we washed the leaf disks three times with 0.05% tween 20 solution before storing the samples. Leaf discs were placed in 1.5 ml tubes, immediately frozen in liquid nitrogen, and stored at -80◦C for downstream analyses.
Aiming to quantify the translocation of applied dsRNA molecules from localized to the systemic leaves, RT-qPCR was performed on the total RNA extracted from the collected samples. Due to the small size of discs collected from leaves, the small-scale RNA isolation protocol from PureLink® Plant RNA Reagent kit (Thermo Fisher Scientific, Waltham, MA) was used for total RNA extraction. Reverse transcription (RT) was carried out with 1 µg of the total RNA in a 20 µl reaction volume using the ProtoScript II First Strand cDNA synthesis kit (NEB, Ipswich – MA, USA). The RNA was denatured at 95◦C for 5 min in the presence of random primer mix and nuclease-free H2O, before adding the remaining RT reagents and conducting the RT steps. Then, 2 µl of cDNA was used for qPCR using the SsoAdvanced Universal SYBR Supermix (BIO-RAD, Hercules-California, USA) in a 10 µl reaction volume, following the manufacturer’s specifications, in a C1000 touch CFX96 real-time PCR system (BIO-RAD, Hercules-California, USA). The qPCR primers were designed to target 250 bp of the central regions of each applied dsRNA (CP, HC-Pro, and NIb) because of the potential degradation of the applied dsRNA in their extremities by exoribonucleases [29], which could mask the assay if primers target the dsRNA extremities. Newly synthesized dsRNAs from each region were added as a template for the positive control in each corresponding reaction, and the mocks (samples from plants not treated with dsRNA) were used as the negative control. The cycle threshold (Ct) values were analyzed to correlate with the dsRNA load in each sample.
Standard curves to assess the DsRNA concentration inside the plant
To estimate the concentration (ng/µl) of dsRNA in leaf tissue, we generated standard curves based on the Ct values obtained from RT-qPCRs run on serial dilutions of the synthesized dsRNA from each genomic region. Aliquots from serial dilutions of dsRNA concentrations (1:10, 1:100, 1:103, 1:104, 1:105, 1:106, 1:107, 1:108, and 1:109) were amplified by RT-qPCR as previously described. A standard curve for each dsRNA was prepared by plotting a linear regression curve with log10 of dsRNA dilutions on the X-axis and Ct values on the Y-axis (n = 3 for each dilution point). Thus, by obtaining the Ct values from the dsRNA translocation experiment, we can enter the data to the Y-axis, from the equation obtained from the linear regression estimated above, and estimate the dsRNA concentration (X) in the sample collected (S2 Fig). Statistical analyses and graph plotting were performed in R [27] using the RStudio interface [30].
Stem-loop RT-PCR to detect VsiRNAs
To investigate whether exogenously applied dsRNAs move in the form of dsRNA or vsiRNAs inside the plant, or how long those molecules remain in the plant, we used the stem-loop reverse transcription PCR (RT-PCR) methodology [31]. This technique is used for detecting and amplifying siRNAs, where the stem-loop is designed in the shape of a hairpin loop and has a 3’ complementary overhang to the siRNA. Based on the vsiRNA profiling analyses described above, a 21-nt vsiRNA was selected from the sense strand of each dsRNAs synthesized (Table 2), for the design of dsRNA-specific stem-loop reverse primers [31]. The vsiRNAs were selected from hotspots, genomic regions with a high abundance of reads with different unique vsiRNA sequences [13], detected on each of the genomic regions where the dsRNAs were designed. For this procedure, total RNA extraction was performed as described above. In the RT step, total RNA (1 µg RNA in 20 µl of reaction) was denatured at 65 ◦C for 5 min in the presence of specific reverse primers for each vsiRNA (Table 3). Then, Protoscript II Reaction Mix and Enzyme Mix were added. To increase the RT efficiency, the pulsed RT-reaction was conducted as follows: 60 cycles of 30 ◦C for 30 s, 42 ◦C for 30 s, 50 ◦C for 1 s, and then 85 ◦C for 5 min to inactivate the enzyme. The PCR conditions were 95 ◦C for 3 min followed by 35 cycles of 95 ◦C for 30 s and 61 ◦C for 1 min. The resultant PCR amplicons were resolved in 2% agarose gel electrophoresis.
Tracking applied DsRNAs with Cy3 fluorophore
To track the movement of dsRNA molecules inside the plant, dsRNA molecules were labeled with cyanine 3-UTP fluorescent dye (Cy3-UTP) by adding 1 µl of Cy3-UTP to the T7 Ribomax reaction. Then, the dsRNA-cy3 was purified using a ChargeSwitch PCR Clean-Up Kit (Thermo Fisher Scientific, Waltham, MA). The dsRNA-cy3 molecules were applied by the same method used in the dsRNA experiment described above. After 24 h, localized (treated) and systemic (untreated) leaves and samples from plant stems were collected to verify the dsRNAs distribution inside the plant. Leaf tissue was prepared using the epidermal peeling method as previously described [32]. Free-hand sections of nonembedded stem tissue were prepared and placed on a slide to detect the Cy3-fluorescence as described previously [33]. All samples were examined using an epifluorescence microscope (AxioImager M1) and the images were processed on ZEN Microscopy Software (Zeiss, White Plains, NY, USA).