Nissl staining assessed neuronal loss after ICH [27]. Specifically, 16 μm-thick coronal frozen brain sections were immersed in Nissl staining solution for 10 min, followed by 2 min of differentiation in graded ethanol. Sections were then mounted and observed under a microscope. Nissl-positive cells were quantified at 20× magnification using ImageJ software (ImageJ 1.5, NIH, USA). H&E staining was performed to evaluate hematoma volume [1]. The section containing the largest hematoma area—typically at the level of the needle track—was selected for staining. The stained sections were scanned using a Leica DM6 microscope, and the hematoma volume was measured using ImageJ software (ImageJ 1.5, NIH, USA).
TUNEL staining
To quantify neuronal apoptosis 72 h after ICH, TUNEL staining (green) was performed using an in situ apoptosis detection kit (C1088, Beyotime Biotechnology, Shanghai, China). TUNEL-positive neurons in the hematoma region were manually counted. Three random sections were analyzed for each brain, and the average number of TUNEL-positive neurons was calculated using ImageJ software (ImageJ 1.5, NIH, USA) at 200× magnification. The results were expressed as the percentage (%) of TUNEL-positive neurons.
Western blot (WB)
Protein samples were mixed with loading buffer and heated at 95 °C for 8–10 min, followed by electrophoresis on a 10% SDS-polyacrylamide gel (E303-01, Vazyme Biotech Co., Ltd., Nanjing, China). Proteins were transferred to methanol-activated PVDF membranes (IPVH00010, Millipore, Billerica, MA, USA). Membranes were blocked with 5% non-fat milk in TBST at room temperature for 2 h and then incubated overnight at 4 °C with primary antibodies (1:1000 dilution). After three washes with TBST (15 min each), membranes were incubated with HRP-conjugated secondary antibodies (1:3000) at room temperature for 2 h. Protein bands were visualized using enhanced chemiluminescence reagents (WBULP-100ML, Millipore, Billerica, MA, USA) and analyzed for band density using ImageJ software (ImageJ 1.5, NIH, USA). Antibody details are provided in Table S2.
RNA isolation and quantitative PCR (qPCR)
Total RNA was extracted from brain tissue 72 h after ICH using Trizol (79306, Qiagen, Hilden, Germany). RNA was reverse transcribed into cDNA using the 5× All-in-One RT MasterMix Kit (R333-01, Vazyme, Nanjing, China). qPCR was performed in triplicate using SYBR Green qPCR MasterMix (Q711-02, Vazyme, Nanjing, China). mRNA expression levels were normalized to β-actin and calculated using the 2-ΔΔCt method. Primer sequences are listed in Table S3.
Immunofluorescence staining
Paraffin-embedded brain tissues were sectioned at a thickness of 4 μm for immunofluorescence staining. Sections were first deparaffinized and rehydrated in xylene and graded ethanol, followed by antigen retrieval in sodium citrate buffer at boiling temperature for 1 h. After preparation, the sections were blocked with 5% BSA for 2 h and then incubated overnight at 4 °C with primary antibodies (1:150 dilution). On the following day, sections were incubated at room temperature for 1.5 h with secondary antibodies—goat anti-rabbit Cy3 (1:200, ab6939, Abcam, USA) and goat anti-rabbit Alexa Fluor 488 (1:200, ab150077, Abcam, USA). Finally, the sections were counterstained with DAPI (C0065, Solarbio, Beijing, China) for 15 min, rinsed, and imaged using a Zeiss fluorescence microscope (Zeiss Axio Imager 2, Germany). Cell immunofluorescence was carried out according to established protocols [21]. Neuronal fibers were quantified by MAP2 immunofluorescence staining, and fiber length was analyzed by tracing with ImageJ software. RAW264.7 or HT-22 cells (3 × 10⁴ cells per well) were seeded on coverslips in 24-well plates and cultured overnight. After drug treatment, the culture medium was removed, and the cells were washed three times with cold PBS. Cells were then fixed with paraformaldehyde for 30 min, permeabilized with 0.5% Triton X-100 for 15 min, and blocked with 5% BSA for 2 h. Fluorescently labeled secondary antibodies were applied and incubated at room temperature for 1.5 h. Cells, stained with DAPI (C0065, Solarbio, Beijing, China) for 15 min, were thoroughly washed and mounted with coverslips. Fluorescence images were captured using a Zeiss Axio Imager 2 microscope (Germany).
Tissue distribution of RVG/FA-NPs@API in ICH model mice
ICH model mice were established and intravenously injected with Cy5.5-labeled RVG/FA-NPs@API (200 µL, 2 mg/mL) via the tail vein 24 h after ICH induction. In vivo fluorescence imaging was performed at 1, 12, and 24 h post-injection using the IVIS Spectrum imaging system (PerkinElmer, USA). Following euthanasia, major organs, including the brain, liver, spleen, lungs, heart, and kidneys, were harvested for ex vivo imaging. Fluorescence intensity was quantified using ImageJ software to analyze the tissue distribution pattern of the nanoparticles.
In vivo targeting evaluation of RVG/FA-NPs@API
After euthanasia, brain tissue was harvested and dissociated into single cells using the Adult Brain Dissociation Kit (130-107−677, Miltenyi Biotec, Germany). CD11b-positive cells were then enriched using CD11b MicroBeads (130-049−601, Miltenyi Biotec, Germany). The enriched cells were subjected to flow cytometry analysis using PE-conjugated anti-mouse CD86 antibodies (105007, BioLegend, USA) at a dilution of 5 µL per 1 million cells in a 100 µL staining volume. Cy5.5 fluorescence was used to evaluate nanoparticle uptake. The proportion of Cy5.5⁺CD86⁺ double-positive cells was analyzed using a BD LSRFortessa™ X-20 flow cytometer (BD Biosciences, USA) to assess the enrichment efficiency of RVG/FA-NPs@API in M1-type macrophages.
Network pharmacology analysis
Network pharmacology analysis was conducted as previously described [28]. We used databases, including SwissTargetPrediction, PharmMapper, and DrugBank, to identify potential targets associated with API. In the SwissTargetPrediction database, targets with a probability score greater than 0.1 were selected as initial candidates. Targets related to ICH were retrieved from DisGeNET, OMIM, and GeneCards. For the GeneCards database, targets with a relevance score of ≥ 20 were included. The keywords used for the search included “intracerebral hemorrhage,” “basal ganglia hemorrhage,” “hemorrhagic stroke,” and “intracranial hemorrhage.” Drug-related and disease-related targets from each database were compared, and duplicate entries were removed. The intersection of the two target sets was identified and visualized using a Venn diagram from the online tool (https://bioinformatics.psb.ugent.be/).
Protein-protein interaction (PPI) network and enrichment analysis
The overlapping targets of the drug and disease were imported into the STRING database, with the organism set to Homo sapiens and the confidence score threshold set to medium (0.4) to generate a PPI network. The resulting interaction data were then imported into Cytoscape software (version 3.10.1), and network topology parameters, including degree, betweenness, and closeness centrality, were analyzed using the CytoHubba plugin. Functional enrichment analysis, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment, was performed using the Metascape database (https://metascape.org/). The screening criteria were set to Homo sapiens and p < 0.05. Final visualizations were generated using the WeChat online charting tool.
Molecular docking
Molecular docking between API and JAK1 protein—including the full-length protein and each of its four individual domains—was performed using AutoDock (version 4.2.6). The protein structure files in PDB format were obtained from the RCSB Protein Data Bank (https://www.rcsb.org/). After removing water molecules from solvents and ligands, hydrogen atoms were added. The PDBQT files of the JAK1 protein and its domains were prepared as receptor files, while the PDBQT file of API was used as the ligand for docking analysis. Docking results were analyzed using AutoDockTools (version 1.5.7), and visualizations were generated using PyMOL (version 2.4.1) and Liplot (version 2.2.8). The root-mean-square deviation (RMSD) between the docked conformations of API and its original structure was calculated using PyMOL. A successful docking result was one with an RMSD ≤ 2.0 Å (0.2 nm).
Molecular dynamics simulation
Molecular docking and dynamics simulations of API with the JAK1 protein and its kinase domain were performed using Discovery Studio (BIOVIA, Dassault Systèmes, Discovery Studio Modeling Environment, version 2019). The molecular dynamics simulation involved the following steps: (1) The protein-ligand complex was imported into Discovery Studio, and the “Prepare Protein” tool was used to preprocess the protein structure. (2) The ligand file was opened, and the CHARMM36 force field was applied for energy minimization of the protein and ligand before simulation. (3) The dynamics process was initiated using the “Standard Dynamics Cascade” protocol, with the solvent system window (Complex.dsv) set as the active window for the molecular dynamics simulation. (4) Hydrogen bond fluctuations within the protein-ligand complex, along with the RMSD and root-mean-square fluctuation (RMSF) values of the protein backbone and side chains, were analyzed using Discovery Studio.
In vitro kinase activity assay
Active human JAK2 protein (Cat# J62-53G, Signal Chem, Canada) was diluted to a final concentration of 0.1 µg/ml in Kinase Dilution Buffer III (Cat# K23-09, Signal Chem, Canada), and then incubated with His-tagged STAT3 substrate (3 µg), purified from E. coli M15 cells using Ni-NTA magnetic beads (Cat# 30210, Qiagen, Germany), together with 5 µl ATP. Different concentrations of apigenin (10 µM and 20 µM; Cat# A800500, Macklin, China) were added to the reaction system, which was maintained at 30 ℃ for 30 min. Reactions were terminated by adding 1× Laemmli buffer (Cat# 1610747, Bio-Rad, USA) and boiling at 95 ℃ for 5 min. Western blotting was performed using an anti-phospho-STAT3 antibody (Cat# 9145, Cell Signaling Technology, USA) to detect JAK2-mediated phosphorylation of STAT3 at Tyr705 [29].
Cell culture and cell viability assay
Cell viability was assessed as previously described [6]. RAW264.7 and HT-22 cell lines (CL-0190/CL-0595, Procell Life Science & Technology Co., Ltd., Wuhan, China) were seeded in 96-well plates at a density of 5 × 103 cells per well in high-glucose DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were treated with various concentrations (0, 12.5, 25, 50, 100, and 200 µM) of API or biotinylated API for 12, 24, and 48 h. After treatment, CCK-8 reagent (C0038, Beyotime Biotechnology, Shanghai, China) was added, and cells were incubated at 37 °C for 2 h in a 5% CO₂ incubator. Absorbance was measured at 450 nm using a spectrophotometer (Thermo Fisher Scientific, USA).
In vitro live/Dead cell staining
Cell apoptosis was assessed using a live/dead cell staining method [30]. According to the manufacturer’s instructions, a Calcein-AM/PI Cell Viability and Cytotoxicity Assay Kit (C2015M, Beyotime, Shanghai, China) was used. Briefly, cultured neurons were washed to remove residual esterases from the medium. A working solution containing Calcein-AM (for viable cells) and Propidium Iodide (PI, for dead cells) was added to the cultured cells, followed by incubation at 37 ℃ for 30 min in the dark. After staining, live/dead cell status was assessed under a fluorescence microscope (Zeiss Axio Imager 2, Germany). The ratio of PI-positive (dead) cells to total cells was quantified to evaluate cell membrane integrity and viability.
Pull-down assay for API-binding proteins
RAW264.7 cell lysates were incubated overnight at 4 °C with biotin, biotinylated API, or biotinylated API pre-incubated with free API. After incubation, streptavidin-conjugated magnetic beads (21115, Thermo Fisher Scientific, USA) were used to pull down proteins from the lysates at 4 °C for at least 6 h. The beads were thoroughly washed with PBS, and 5× loading buffer was added. Samples were boiled, and the supernatant was collected to detect JAK1 protein.
Surface plasmon resonance (SPR)
The binding affinity between API and JAK1 protein (HY-P700583, MedChemExpress, USA) was evaluated using the SPR-based Biacore T200 instrument (Cytiva, Sweden). A CM5 sensor chip was used for the experiment. The sensor surface was activated with a mixture of 50 mM NHS and 200 mM EDC for 7 min. JAK1 protein (420 µg/mL), diluted in 10 mM acetate buffer (pH 4.5), was immobilized on the chip surface at a 10 µL/min flow rate. The surface was then blocked with 1 M ethanolamine (pH 8.5). Multiple binding cycles were performed, and response signals were recorded with time on the x-axis and response units on the y-axis. The collected data were fitted using the Biacore T200 evaluation software with a 1:1 Langmuir binding model to determine kinetic parameters, including the association rate constant, dissociation rate constant, and equilibrium dissociation constant. The experimental procedures and data analysis were carried out by TGTMED Pharmaceutical Technology Co., Ltd. (Shanghai, China).
Cellular thermal shift assay (CETSA)
CETSA was performed as previously described [31]. Briefly, ICH mice were intraperitoneally injected with API (20 mg/kg), and RAW264.7 cells were treated with API (25 µM) for 8 h. After treatment, brain tissue from the hemorrhagic side of mice and protein lysates from RAW264.7 cells were collected. Protein stability was assessed at temperatures ranging from 50 °C to 71 °C. Following thermal denaturation, samples were subjected to three freeze-thaw cycles using liquid nitrogen, then centrifuged at 20,000 rpm for 20 min at 4 °C. The supernatants were collected for further analysis.
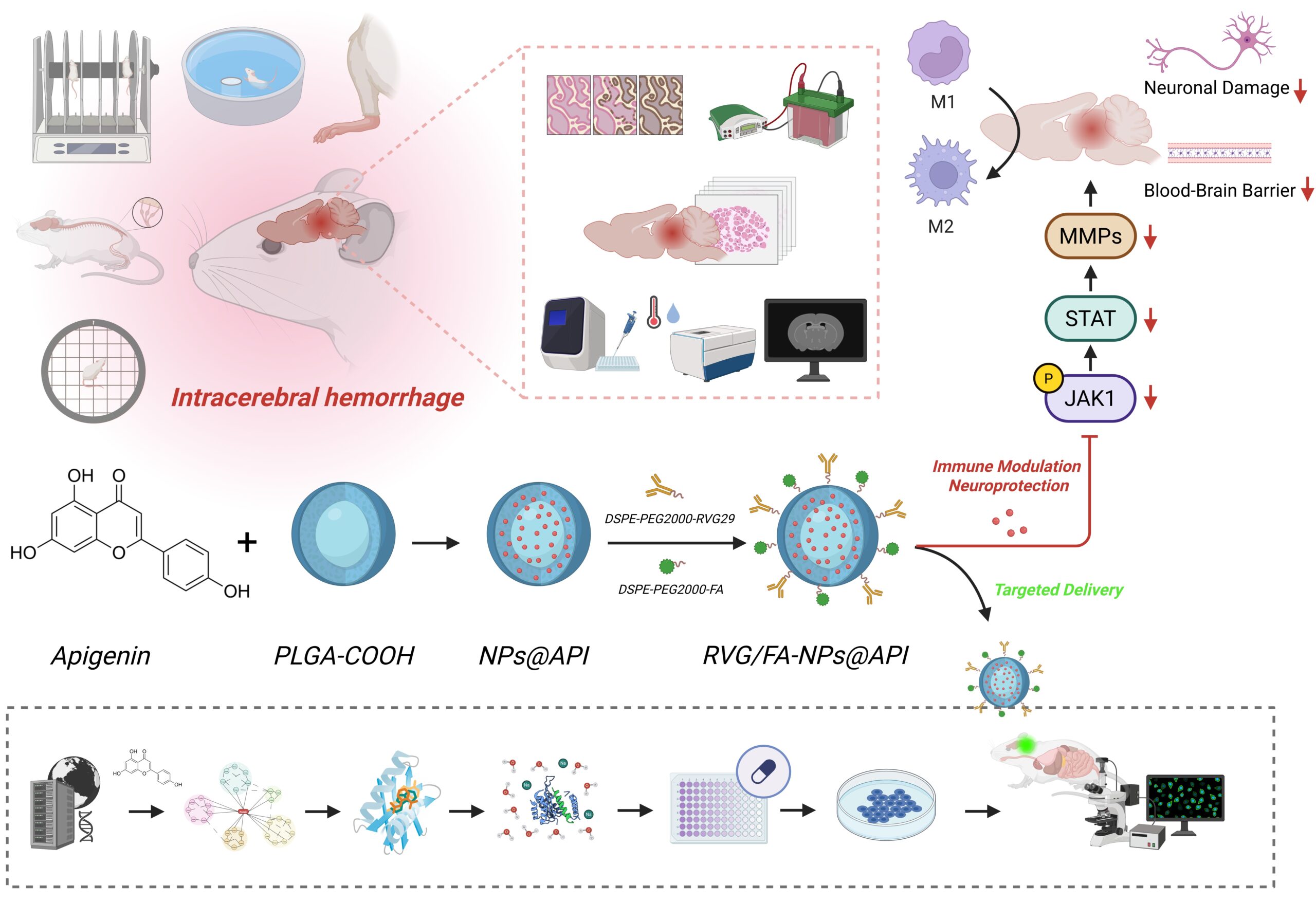
Preparation of PLGA nanoparticles and API loading
API-loaded PLGA nanoparticles were prepared using the solvent evaporation method. Briefly, 50 mg of PLGA-COOH (LA: GA = 50:50, Mw = 30,000–60,000; P2191, Sigma-Aldrich, USA) was dissolved in 4 mL of dichloromethane (DCM; D807825, Macklin, China), followed by the addition of 10 mg of API. The mixture was thoroughly stirred to form the organic phase. This solution was then added dropwise into 40 mL of an aqueous phase containing 1% polyvinyl alcohol (PVA, Mw = 30,000–70,000; P8136, Sigma-Aldrich, USA) under ice bath conditions. A probe-type sonicator (Scientz-IID, Ningbo Scientz Biotechnology Co., Ltd., China) was used to ultrasonicate the mixture at 200 W for 3 min (2-second pause, 1-second pulse). The resulting emulsion was stirred at 500 rpm under ventilation for 4 h to allow DCM evaporation. The formed nanoparticles were collected by centrifugation at 12,000 rpm for 10 min (Model: 5810R, Eppendorf, Germany), washed three times with deionized water, and freeze-dried for storage [32, 33].
Surface functionalization with RVG29 and FA
Dual modification was achieved by incorporating DSPE-PEG2000-RVG29 (HY-172705, MedChemExpress, USA) and DSPE-PEG2000-FA (PS2-DEFA, Pengshuo Biotech, China). Briefly, 5 mg of lyophilized PLGA nanoparticles were dispersed in 10 mL PBS (pH 7.4), followed by the addition of 0.5 mg DSPE-PEG2000-RVG29 and 0.5 mg DSPE-PEG2000-FA. The mixture was sonicated in a 37 °C water bath for 10 min and stirred at room temperature for 12 h. The modified nanoparticles were washed three times by centrifugation at 10,000 rpm, lyophilized, and designated as RVG/FA-NPs@API [34]. To evaluate the respective targeting roles of RVG29 and FA, single-modified nanoparticles were also prepared, namely RVG-NPs@API and FA-NPs@API. The procedure was identical to that of the dual-modified group, except that only 0.5 mg DSPE-PEG2000-RVG29 or 0.5 mg DSPE-PEG2000-FA was added, followed by lyophilization for storage.
Characterization of particle Size, zeta potential, and morphology
Nanoparticle morphology and size were characterized using transmission electron microscopy (TEM) and dynamic light scattering (DLS). TEM: Approximately 3 µL of nanoparticle suspension was dropped onto a carbon-coated 200-mesh copper grid, allowed to stand at room temperature for 5 min, excess liquid removed, and negatively stained with 3 µL of 1% (w/v) uranyl acetate for 5 min. After drying, samples were observed at 80 kV using a TEM (Tecnai-10, Philips, The Netherlands). DLS: Hydrodynamic diameter, polydispersity index (PDI), and ζ-potential were measured using a Zetasizer Nano ZS90 (Malvern Instruments, UK) with three independent replicates [35]. Structural Confirmation: RVG/FA-NPs@API (6 mg) was dissolved in DMSO-d6/D2O (3:2) and analyzed by 1 H NMR spectroscopy to verify conjugation [36].
X-ray diffraction (XRD)
XRD patterns were obtained using a diffractometer equipped with a Cu target and graphite monochromator (Rigaku D/max 2500/PC, Japan). Cu-Kα radiation (λ = 1.54 Å) was used as the incident beam, with a scanning range of 5°−90°, operating at 40 kV and 200 mA, and a scan rate of 5°/min with a 1-s counting time.
Determination of drug encapsulation efficiency (EE%) and loading efficiency (LE%)
To evaluate the drug EE%, 5 mg of freeze-dried RVG/FA-NPs@API was dissolved in 1 mL of acetonitrile by sonication. The concentration of API was then determined using a high-performance liquid chromatography (HPLC) system (Agilent 1260, USA). A C18 column (4.6 mm × 250 mm, 5 μm; Waters, USA) was used with a mobile phase of methanol: water (60:40, v/v) at a 1.0 mL/min flow rate. The detection wavelength was set at 340 nm. EE% and LE% were calculated based on the standard calibration curve [34].
Stability evaluation
To assess the stability of RVG/FA-NPs@API, the nanoparticles were incubated in PBS buffer and cell culture medium containing 10% fetal bovine serum (FBS) for 0 and 24 h. At the designated time points, aliquots of the nanoparticle suspension were collected, and particle size was measured using DLS. For long-term storage stability evaluation, freshly prepared RVG/FA-NPs@API were stored at 4 °C in the dark. Samples were taken on days 0, 15, 30, 45, and 60, and particle size was measured via DLS to assess changes over time.
In vitro drug release study
A total of 5 mg of RVG/FA-NPs@API was resuspended in 1 mL of PBS (containing 0.5% Tween-80, at either pH 7.4 or pH 5.5) and placed into a dialysis bag (MW cut-off 10,000 Da; Spectrum Labs, USA). The bag was then immersed in 50 mL of the corresponding buffer and incubated at 37 °C with constant shaking at 100 rpm. At 1, 2, 5, 7, 10, 25, and 50 h, 1 mL of the release medium was collected and replaced with an equal volume of fresh buffer. The concentration of API in the release samples was determined using HPLC.
In vitro biocompatibility assessment of nanoparticles
RAW264.7 cells were seeded in 6-well plates at a density of1 × 10⁵ cells/well and polarized into M1 macrophages by stimulation with LPS (1 µg/mL, L4391, Sigma-Aldrich, USA) for 12 h, or into M2 macrophages by stimulation with IL-4 (20 ng/mL, 200-04, PeproTech, USA) for 24 h [37]. Polarized RAW264.7 cells and HT22 cells were then seeded into 96-well plates (5 × 10³ cells/well) and cultured in high-glucose DMEM supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37 ℃ in a 5% CO2 incubator. Cells were treated with RVG/FA-NPs@API at different concentrations (5–25 µg/mL) for 24 h, followed by the addition of 10 µL/well CCK-8 reagent (Dojindo, Japan). After incubation for 2 h at 37 °C, absorbance was measured at 450 nm [38].
In vitro cellular uptake of nanoparticles
RAW264.7 cells were seeded into 6-well plates at a density of 2 × 10⁵ cells per well and incubated at 37 °C in a 5% CO₂ atmosphere for 24 h. The cells were then transferred to a fresh DMEM medium containing FITC-labeled RVG/FA-NPs@API (FITC conjugation achieved via covalent grafting; Sigma-Aldrich, USA) and incubated for 6 h. After incubation, all cells were fixed with 4% paraformaldehyde for 20 min and washed with PBS. The nuclei were stained with DAPI for 15 min. Fluorescence images were captured using a confocal laser scanning microscope (CLSM), and cellular uptake of RVG/FA-NPs@API was quantitatively analyzed using a flow cytometer (BD FACSvantage SE, USA) [39].
Statistical analysis
All experiments were conducted in a randomized and blinded manner. Each in vitro experiment was independently repeated at least three times, and each In vivo group included more than three mice. Statistical analyses were performed using GraphPad Prism 10.0 software (GraphPad Software Inc.). For comparisons between two groups, Tukey’s multiple comparison test was used. For multiple group comparisons, one-way or two-way analysis of variance (ANOVA) was applied, followed by appropriate post hoc tests. A p-value < 0.05 was considered statistically significant. All data were expressed as mean ± standard deviation (SD).