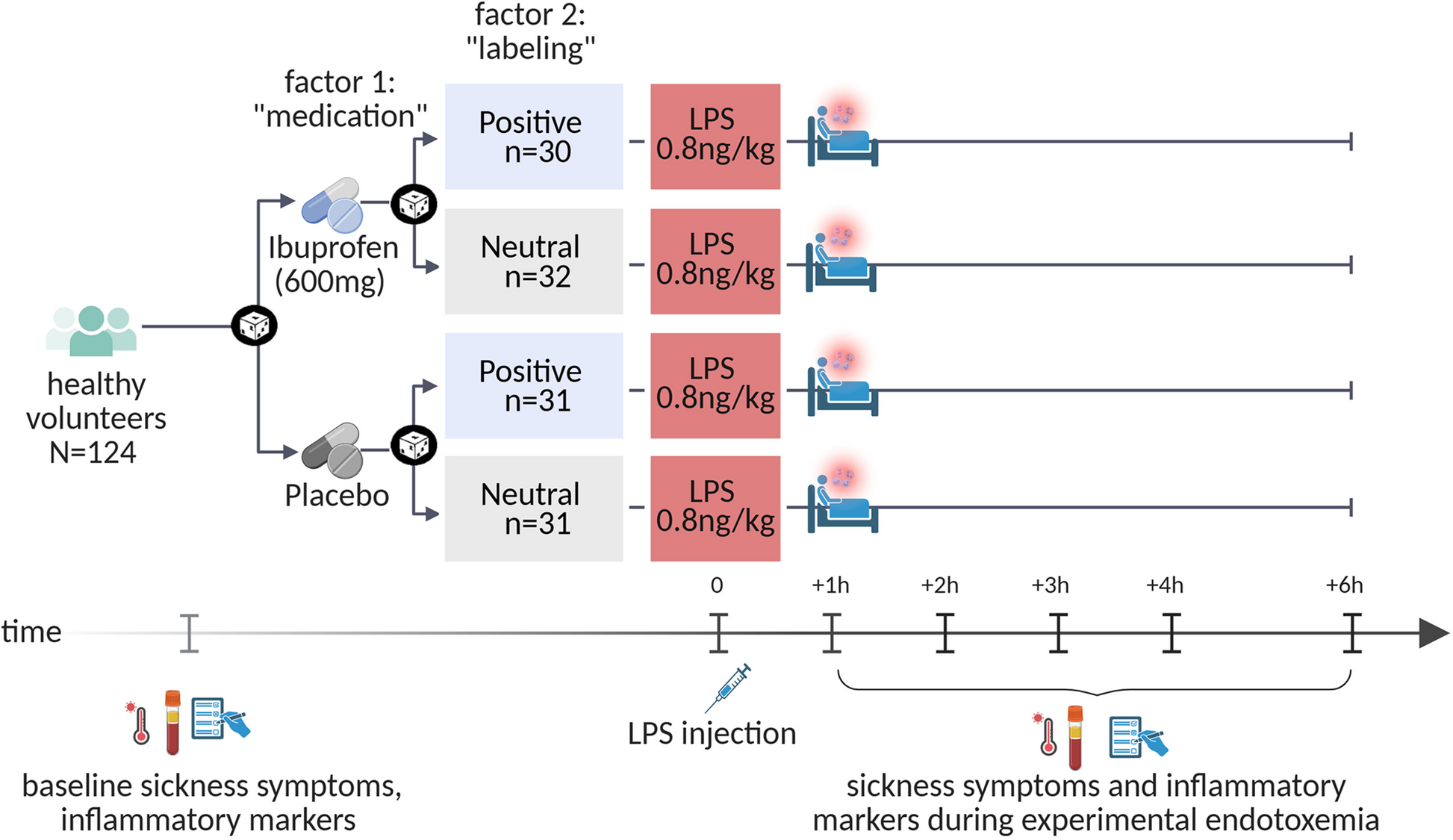
Aim, design and setting of the study
In this randomized, placebo-controlled, double-blind experimental study, all N = 124 participants received an intravenous injection of LPS (0.8 ng per kg of body weight, details on endotoxemia below) to induce acute systemic inflammation along with sickness symptoms, following established protocols [19, 20].
The study aim was to examine the main and interaction effects of positive treatment expectations and anti-inflammatory medication. To this end, we implemented a fully balanced 2 × 2 factorial study design [4] (see Fig. 1). Participants were randomized to receive a single oral dose of either 600 mg ibuprofen or an inert substance (placebo) prior to LPS administration. The administration of ibuprofen or placebo was randomly paired with standardized verbal labeling delivered by the study physician to induce either positive or neutral expectations. This resulted in four experimental groups:
-
1.
Ibuprofen combined with positive labeling (IBU + pos)
-
2.
Ibuprofen combined with neutral labeling (IBU + neu)
-
3.
Placebo capsule combined with positive labeling (PLA + pos)
-
4.
Placebo capsule combined with neutral verbal labeling (PLA + neu)
Study design. All volunteers were intravenously injected with endotoxin (LPS: 0.8 ng/kg of body weight) to induce acute systemic inflammation. In a 2 × 2 factorial between-group design, volunteers were randomized to receive ibuprofen or placebo (factor 1 “medication”) prior to injection, randomly combined with positive or neutral verbal treatment information delivered by the study physician (factor 2 “labeling”). Outcome measures (i.e., bodily and affective sickness symptoms, inflammatory, neuroendocrine and vital parameters) were assessed prior to LPS injection (i.e., baseline) as well as 1, 2, 3, 4, and 6 h after injection. Created with BioRender.com
The 2 × 2 factorial, balanced placebo design allows the investigation of the effects of physician communication (main effect of labeling), anti-inflammatory medication (main effect of medication), and their interaction (labeling × medication interaction) on outcomes.
The study was conducted at the University Hospital Essen, University of Duisburg-Essen, Germany, as part of the Collaborative Research Center (CRC) 289 “Treatment Expectation”, funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG). The overarching goal of the CRC is to elucidate mechanisms and clinical implications of treatment expectations. This study was accomplished as part of subproject A11 (PIs: authors S.B., H.R.), which addresses expectancy effects in the context of inflammation. The work was conducted in accordance with the Declaration of Helsinki, approved by the local Institutional Ethics Review Board (19–8755-BO), and preregistered in the German Clinical Trials Register (DRKS) on October 22nd 2020 (before inclusion of the first participant on March 2nd, 2021), registration number DRKS00023088; for further details, see trial registration website: German Clinical Trials Register. The manuscript adheres to the CONSORT guidelines and BMC’s editorial policies. All participants provided written informed consent and received financial compensation for their participation, which involved multiple visits before and after the study day for screening and safety purposes. Study procedures included the administration of LPS and the assessment of outcomes, such as inflammation-related markers derived from blood samples and sickness symptoms (reported herein). Additional experimental outcomes were acquired using a modified Posner task [21] for measures elucidating the influence of emotional distractors on spatial attention and algometry for pressure pain sensitivity [22], which will be reported elsewhere given our focus on patient-reported outcomes relevant to sickness symptoms herein. Further assessments were conducted as part of the CRC’s central projects, which coordinate and administrate standardized collection of additional measures across all study sites and subprojects involving human participants for merged analyses (for a preregistration, see: OSF Registries | SFB289—Central Project NeuroImaging). These data include brain imaging assessment of structural and functional brain connectivity prior to experimental study protocols; assessment of cortisol awakening response and salivary alpha amylase activity in saliva; and a comprehensive questionnaire battery. Embedded within the larger CRC approach, these measures will serve analyses across projects aiming to identify predictors of expectancy effects, along with additional psychological trait and state measures related to stress, negative affectivity, and pain, as accomplished for example herein [23]. For the purposes of sample characterization herein, we report sociodemographic characteristics and trait anxiety and depression measured with the trait version of the validated German State-Trait Anxiety Depression Inventory (STADI) [24].
Participants
Healthy men and women aged 18–45 years were newly recruited for this study through public advertising. All volunteers received detailed information about the study and underwent a highly standardized and well-established medical examination and screening process as implemented in our previous studies (e.g., [16, 19, 20]). This process included a thorough medical history, physical examination, and repeated assessments of laboratory parameters [blood cell count, liver enzymes, renal retention parameters, coagulation factors, C-reactive protein (CRP)]. General exclusion criteria included a history and/or current signs of gastrointestinal, hepatic, renal, cardiovascular, hematologic, or respiratory disease; obesity, diabetes mellitus, or other metabolic disorders; abnormal laboratory values; regular use of medications, especially analgesics; any contraindication to ibuprofen use; evidence of depression and anxiety based on the validated Hospital Anxiety and Depression Scale (HADS); MRI-specific exclusion criteria (phobic anxiety, claustrophobia, ferromagnetic implants, etc.); pregnancy or lactation. On the study day, women were tested for pregnancy using a urine hCG test. Sex assigned at birth was collected as self-report (male, female, diverse). As the study was conducted during the COVID-19 pandemic, additional safety measures were implemented in cooperation with the Department of Infectious Diseases, University Medicine Essen. These measures ensured the protection of study participants and personnel, and met the requirements of the health authorities. They included repeated COVID-19 testing and a mandatory six-week interval between the participant’s last COVID-19 vaccination (or last symptom of a coronavirus infection) and study entry.
Note that the originally planned sample size of N = 168 could not be achieved due to a 9-month delay in study start caused by the COVID-19 pandemic. For the current sample size of N = 124, post hoc power analysis using G*Power (version 3.1.9.7) indicated a sufficient statistical power (1-β = 0.98) to detect small to medium-sized interaction effects (f = 0.15, α < 0.05) in the primary outcomes. All raw data are provided in Additional file S1.
Experimental endotoxemia
Lyophilized LPS (reference standard endotoxin from Escherichia coli O113:H10, lot H0K354, United States Pharmacopeia, Rockville, MD, USA) was prepared for human use as previously described [19] and underwent microbial safety testing at a cGMP-certified laboratory (Labor LS, Bad Bocklet, Germany). On the study day, an intravenous catheter was placed in an antecubital forearm vein for repeated blood withdrawals and LPS injection. All volunteers were injected with a dose of 0.8 ng LPS per kg of body weight, which has been demonstrated by our and other groups [20, 25, 26] to reliably induce a systemic immune activation and sickness symptoms. Participants were instructed to refrain from strenuous physical activity for 48 h before and on the day of the study. For safety, volunteers were continuously monitored by the study physician for 6 h after LPS injection and returned to the laboratory for follow-up visits 24 h and 1 week after injection.
Notably, the study did not include a non-LPS condition, as its primary objective was to examine the effects of psychological (labeling) and pharmacological (ibuprofen) interventions on LPS-induced symptoms. The effects of LPS versus saline injections have been extensively analyzed by our and other research groups across all study outcomes, consistently demonstrating no or negligible changes in inflammatory parameters and sickness symptoms in non-LPS (saline) conditions [16, 19, 20, 25, 26].
Randomization and masking
Randomization for ibuprofen versus placebo and positive versus neutral labeling was conducted using an online tool (www.randomizer.org) by a person not involved in the study. The study physician received a sealed envelope containing the identical-looking ibuprofen or placebo capsule (blinded) as well as an instruction regarding the treatment-related labeling. The ALIIAS software tool [27], developed and utilized within the CRC, was employed to generate a pseudonym allowing sharing and integration of data into the CRC’s data cloud.
Anti-inflammatory or inert medication
Forty-five minutes prior to the LPS injection, volunteers received either the anti-inflammatory drug ibuprofen (1 capsule, 600 mg per os) or an identically appearing but inert capsule as a placebo, administered in a randomized, double-blind manner. The choice of drug and dosage was informed by evidence that ibuprofen (i.e., 3 × 800 mg/day) effectively reduces fever, headache, and myalgia in previous endotoxemia studies utilizing significantly higher doses of LPS (i.e., 2–4 ng per kg of body weight) [28]. The timing of drug administration was selected to ensure that the pharmacological effect coincided with the LPS injection, accounting for the delayed onset of action due to the encapsulation of the ibuprofen active ingredient. Encapsulation was necessary to maintain blinding by fabrication of a capsule identical in appearance to the inert substance, as provided by the University Hospital’s central pharmacy.
Positive or neutral labeling
To augment positive treatment-related expectations, the administration of ibuprofen or placebo was randomly paired with a positive (vs. neutral as a control) labeling delivered by the study physician (see Fig. 2). In the positive labeling condition, participants were informed: “You are receiving the well-established anti-inflammatory drug ibuprofen which has been shown in previous studies to effectively improve sickness symptoms”. To reinforce this information, elements of the positive labeling were repeated by the study physician during blood sampling as booster statements (after symptom ratings to avoid directly influencing symptom reporting).
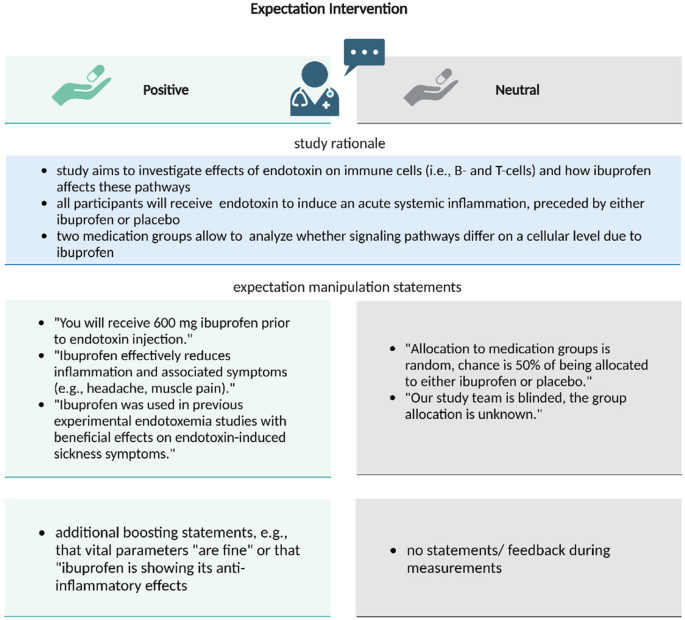
Overview of the labeling conditions: The administration of ibuprofen or placebo was randomly paired with a positive (vs. neutral as a control) labeling delivered by the study physician to augment positive treatment expectations (Positive) vs. a control condition (Neutral) based on distinct treatment information delivered by the physician. Elements of the positive labeling were repeated as booster statements. Created with BioRender.com
In the neutral labeling control condition, participants were informed: “You will receive either an inert substance (“sugar pill”) for control purposes or ibuprofen, with a 50%/50% chance as is usual in experimental settings”.
The positive and neutral treatment communication was scripted and delivered in a standardized manner. Adherence to the protocol was evaluated by an independent observer as previously described [13]. Similar procedures and instructions have been successfully used by our group [13].
Outcomes
Sickness symptoms
Self-reported bodily and affective sickness symptoms were assessed with validated questionnaires at baseline and 1, 2, 3, 4, and 6 h after LPS injection (see study design, Fig. 1) following established procedures (e.g., [19, 20]). All questionnaires were completed digitally with the open-source application LimeSurvey (LimeSurvey GmbH, Hamburg, Germany).
The number and intensity of bodily sickness symptoms were evaluated with an adapted version of the Generic Assessment of Side Effects Scale (GASE). Briefly, volunteers rated the severity of 23 physical symptoms on a 4-point Likert scale (0 = “not present”, 1 = “mild”, 2 = “moderate”, 3 = “severe”) as previously accomplished (e.g., [19]). For the calculation of the sum score, symptoms were included only if volunteers attributed the symptoms to endotoxin administration (endotoxin-attributed total score), reflecting the severity of symptoms associated with endotoxin exposure. The resulting sum score was used for analysis, with higher scores indicating greater bodily sickness. Twenty-four hours after the LPS injection, volunteers were asked to rate their overall symptom burden retrospectively using the GASE.
For inflammation-induced changes in affective symptoms, we implemented the state version of the STADI [24]. It comprises 20 Likert-scaled items ranging from 1 (“not at all”) to 4 (“very much”). The 10-item subscale “Anxiety” comprises the affective (agitation) and cognitive (apprehension) dimensions of anxiety. The 10-item subscale “Depression” measures positive (euthymia) and depressed/negative (dysthymia) mood. The “Global score” combines the anxiety and depression subscales, with higher scores indicating more pronounced negative affectivity.
Additional measures of mood-related symptoms of sickness and fatigue, which were previously established in the context of LPS studies [19, 20, 22], were used to validate and refine the results. These measures included the State-Trait Anxiety Inventory (STAI, state version) to measure state anxiety [29]; the Multi-Dimensional Mood Questionnaire (MDBF, subscale positive mood) to assess changes in positive mood [30]; and the Karolinska Sleepiness Scale (KSS) to indicate fatigue [31, 32]. Affective and fatigue symptoms were also measured 24 h after LPS injection to ensure a return to baseline and as a safety measure. For more information, see the Additional file 2, sections S1 and S2.
Inflammatory, neuroendocrine and vital parameters
To document the time course and extent of LPS-induced systemic inflammation as well as the effects of the interventions, blood samples were collected before (baseline) and 1, 2, 3, 4, and 6 h after LPS injection. Blood samples were collected in ethylenediaminetetraacetic acid (EDTA) treated cubes (7.5 ml S-Monovette® EDTA, Sarstedt AG, Nürnbrecht, Germany), immediately separated by centrifugation (10 min, 4 °C, 2000 × g), and stored at −80 °C until analysis. Plasma concentrations of Tumor necrosis factor-α (TNF-α) and Interleukin (IL)−6 were measured using commercial enzyme-linked immunosorbent assays (ELISA; Quantikine® IL-6/Quantikine® high-sensitivity TNF-α ELISA; R&D Systems, Minneapolis, Minnesota) according to the manufacturer’s protocol and quantified with a Fluostar OPTIMA Microplate Reader (BMG Labtech, Offenbach, Germany). The minimum detectable dose (MDD) as an indicator of the assays’ sensitivity was 0.70 pg/mL for IL-6 and 0.022 pg/mL for TNF-α. As an indicator of hypothalamic–pituitary–adrenal (HPA)-axis activity, plasma levels of adrenocorticotropic hormone (ACTH) and cortisol were measured using commercial ELISA (IBL International, Hamburg, Germany). The MDD was 4.03 ng/mL for cortisol and 1 pg/mL for ACTH. Body temperature was measured using an auricular thermometer (GeniusTM tympanic thermometer), Covidien, Mansfield, USA) and heart rate with an automated device (Omron HBP-1300, Omron Healthcare Co. Ltd., Kyoto, Japan).
Statistical analyses
Statistical analyses were performed with SPSS (Statistical Package for the Social Sciences), Version 29.0.2.0 (IBM Corp., IBM SPSS Statistics for Windows, Armonk, NY, USA) and G*Power (Version 3.1.9.7). Data were plotted using GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA). In case of significant deviations from normal distribution (i.e., IL-6, TNF-α, ACTH, cortisol), logarithmic transformations were applied for statistical analysis. For visualization purposes, plotted results show untransformed data.
Sociodemographic and psychological variables in the experimental groups were compared using univariate analysis of variance (ANOVA) or chi2-tests (for dichotomous variables). For primary and secondary outcomes, repeated measures (rm) ANOVAs were computed with the within-subject factor “time” (i.e., repeated assessments of outcomes at the experimental day at 6 time points) and the between-subjects factors “medication” (ibuprofen, placebo) and “labeling” (positive, neutral). In case of violation of the assumption of sphericity, Greenhouse–Geisser correction was used, and corrected degrees of freedom (df) are reported. Significant main or interaction effects were followed up in secondary analyses: To test our directed hypotheses that positive labeling improves outcomes, positive and neutral labeling groups were separately compared within the medication and within the placebo pill conditions, respectively. For subgroup comparisons, one-tailed independent t-tests were used at single time points (with Bonferroni correction for multiple comparisons of subgroups). Primarily for visualization purposes in figures, changes in symptom scores from baseline to the peak of inflammation (i.e., 3 h post-injection) were computed as delta of peak minus baseline and compared with independent t-tests. Higher positive change scores indicate a greater increase in symptoms. Finally, additional correlational and regression analyses were conducted to explore whether inflammation-induced affective symptoms were related to inflammatory markers and/or to bodily symptoms (reported in Additional file 2, section S3). Alpha level was set at 0.05 unless otherwise indicated, and results are shown as mean ± standard error of the mean (SEM).
Role of the funding source
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation: project-ID 422744262–TRR 289). The funder of the study had no role in study design, data collection, data analysis, or writing of the report.