Structural analysis
The geometrical parameters of the host molecules, [6]CPPAs and [6]CPPDs, and their corresponding host-guest complexes have been meticulously analyzed to elucidate the structural attributes governing their interactions with various guest molecules, carbon nanotubes, and fullerenes (C60 and C70). These analyses are crucial for optimizing the design and functionality of these supramolecular assemblies. As previously designed, the optimized structures of both [6]CPPAs and [6]CPPDs were re-optimized, as shown in Figure. 2. The optimized structures of [6]CPPAs and [6]CPPDs reveal distinct geometrical differences, primarily driven by the substitution of acetylene (C ≡ C) units in [6]CPPAs with azobenzene (N = N) units in [6]CPPDs. This substitution deviates from perfect circularity in the molecular rings, particularly affecting the long-axis measurements. In [6]CPPAs, the long axis is measured at 13.25 Å, whereas in [6]CPPDs, it is slightly reduced to 12.15 Å, reflecting the increased rigidity and reduced conformational flexibility associated with the nitrogen-nitrogen bond in [6]CPPDs. Similarly, the short-axis measurements further corroborate the structural impact of the azobenzene substitution, with [6]CPPAs having a short axis of 12.89 Å and [6]CPPDs slightly lower at 11.81 Å. These dimensions indicate that due to their planar nature and steric bulk, the azobenzene units cause a slight contraction of the ring structure in [6]CPPDs, resulting in a more elliptical and less circular geometry than [6]CPPAs.
Optimized structures of [6]CPPAs (in red color) and [6]CPPDs (in blue color) at M06-2X/6-31G(d).The long and short axes in Å are shown in the Figure.
A detailed analysis by Ali et al. shows the importance of understanding the strain energy (SE) and enthalpy of the formation of [6]CPPDs19. Their finding is of utmost importance in explaining the slight structural distortions observed in [6]CPPDs compared to [6]CPPAs. It helps contextualize the geometrical differences between these molecules, underlining the significance of our research in supramolecular chemistry.
To validate the accuracy of these geometrical observations, the findings were rigorously compared with previous computational and experimental data, which consistently aligned with established trends in guest encapsulation efficiencies and intermolecular distances41. The detailed analysis of critical geometrical parameters, including radius, long axis, and short axis, provides a comprehensive understanding of how the structural modifications between [6]CPPAs and [6]CPPDs influence their ability to interact with and encapsulate guest molecules are tabulated in Supporting Information Table S4. These optimized geometries serve as the basis for designing host-guest complexes, where each guest molecule is individually optimized under the same computational level as the hosts (Fig. 3). The study reports that the cavity size of [6]CPPAs, approximately 13.3 Å in diameter, is nearly ideal for encapsulating C60, while [6]CPPDs, with a slightly smaller cavity, still effectively encapsulates C60 but with a slightly altered binding energy and interaction dynamics.
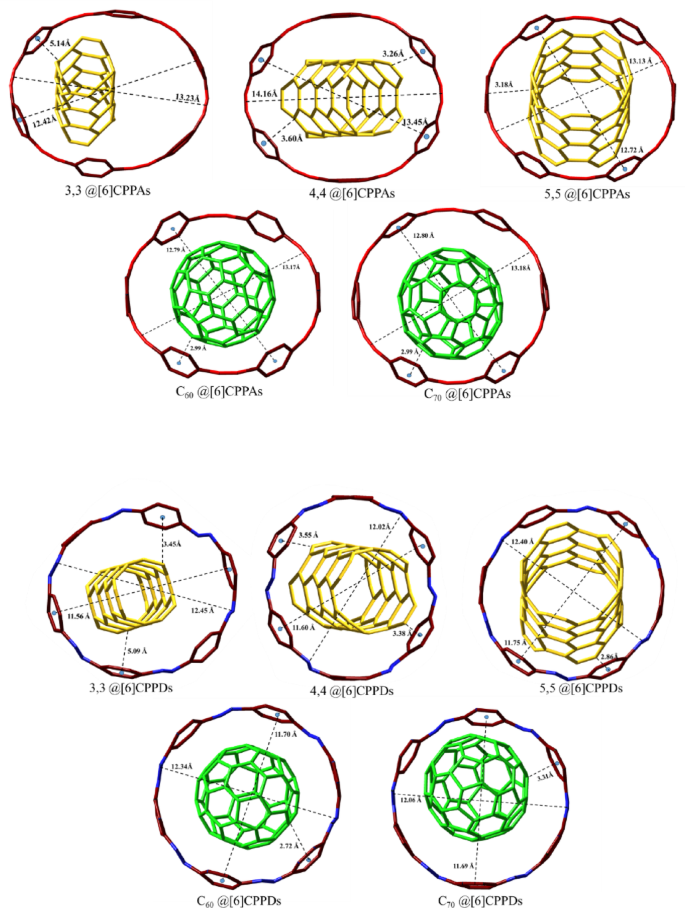
The optimized geometries of host-guest interaction for [6]CPPAs and [6]CPPDs. The long and short axes in Å are shown in the Figure.
The geometry optimization of guest systems, including fullerenes (C60 and C70) and CNTs, reveals that the host’s structural parameters largely govern their encapsulation within the host molecules. The experimental diameters of CNT(5,5) and fullerene (C70) are consistent with the optimized values, falling within 6.6-6.9 Å and 7-7.2 Å, respectively (6.80 Å and 7.01 Å)46,47. The study by Zhao et al. emphasizes the importance of size selectivity in these interactions, where the alignment and overlap of π-systems play a crucial role in the strength of the host-guest binding25. The interfacial length, defined as the distance between the centroids of the phenyl rings of the host and the nearest carbon atoms of the guest, emerges as a critical determinant of interaction strength. For instance, in the C60@[6]CPPAs complex, the interfacial length is measured at 3.1 Å, indicative of π-π interactions. In contrast, the C60[6]@CPPDs complex exhibits a slightly longer interfacial length of 3.2 Å. An important finding from the nano-Saturn study, a seminal work in molecular encapsulation, is the relationship between the interaction angle and binding energy48. The study notes that the closer the angle between the plane of the host and the guest molecule is ~ 90°, the stronger the π-π interactions, leading to more incredible binding energy. This is relevant for the [6]CPPAs and [6]CPPDs systems, where the orientation of the guest molecule within the host cavity can significantly influence the stability and strength of the interaction.
The distance between the centers of the host and guest molecules (dc-c) is another critical parameter that provides insight into the binding affinity and structural alignment within these complexes. In [6]CPPAs-based complexes, particularly those involving (5,5)CNT and fullerene C60, the dc-c distances are notably shorter, indicating a robust binding affinity and efficient encapsulation of the guest molecules. Conversely, in [6]CPPDs-based complexes, the dc-c distances are slightly longer, suggesting a less intimate interaction, which may affect the overall stability and strength of the host-guest complex. These variations in dc-c distances underscore the importance of precise geometrical alignment in determining the effectiveness of molecular encapsulation and the resulting interaction strength.
The analysis of size selectivity within the host molecules further reinforces the differences between [6]CPPAs and [6]CPPDs. For example, among the CNT guests within [6]CPPAs, the CNT(5,5) configuration exhibits the closest binding affinity, followed by CNT(4,4) and CNT(3,3), highlighting the size-selective nature of the [6]CPPAs ring. In contrast, within the [6]CPPDs host, the CNT(4,4) guest is optimally encapsulated, while more prominent guests such as CNT(5,5) and C60 demonstrate significant embedding, as evidenced by minimal dc-c distances indicative of near-complete encapsulation. Notably, the complexation of C70 within [6]CPPDs adopts a “ball in a bowl” configuration, where the guest molecule is positioned above the host ring, leading to limited interaction compared to the more intimate and pronounced interactions observed in the [6]CPPAs counterpart. This configuration suggests that [6]CPPDs may be better suited for guest molecules not requiring complete encapsulation, potentially due to the ring strain and steric effects introduced by the azobenzene substitution. The broader interfacial distances observed in the C70@[6]CPPDs complex further support this interpretation, indicating that the azobenzene units may disrupt the ideal π-π stacking interactions necessary for strong host-guest binding. Our findings align with established trends in guest encapsulation efficiencies and intermolecular distances, reinforcing the robustness of our computational approach and affirming the reliability of predicted host-guest interactions. The complete encapsulation of the smaller molecule, HMB, in both [6]CPPAs and [6]CPPDs complexes is a concrete demonstration of their effective size discrimination capabilities without inducing structural deformations (see the Supporting Information Table S5 and Table S6).
Thermodynamic and kinetic stability
The thermodynamic data regarding the encapsulation of CNTs, C60, and C70 by the hosts, computed at the M06-2X/6-31G(d) level of theory, are tabulated in Table S7 of Supporting information and shown in Fig. 4. The analysis of ΔG(298 K) and ΔS values alongside ΔEcp. values reveals a consistent pattern, underscoring the meticulousness and reliability of the thermodynamic parameters analyzed. The results obtained for complexes involving [6]CPPAs are in close agreement with previously reported findings, affirming the robustness of the M06-2X optimization with dispersion correction. In CNT@[6]CPPAs complexes, all CNT species exhibit heightened stability and adequate size selectivity within the host ring. The (3,3)@[6]CPPAs complex exhibits the weakest binding (ΔE = −20.8 kcal/mol) among them. The stability improves significantly in the (4,4)@[6]CPPAs complex (ΔE = −31.17 kcal/mol), indicating that a slight increase in nanotube diameter enhances interaction strength. The (5,5)@[6]CPPAs complex exhibits the strongest interaction (ΔE = −42.39 kcal/mol), confirming that larger CNTs achieve better stabilization within the CPPA ring. The increasing negative entropy values (ΔS) across these complexes further support the idea that host-guest interactions stabilize the system while restricting configurational flexibility. In contrast, CNT encapsulation within [6]CPPDs follows a different binding pattern due to the structural rigidity and electrostatic contributions introduced by nitrogen incorporation. The (3,3)@[6]CPPDs complex exhibits moderate stability (ΔE = −27.37 kcal/mol), indicating that electrostatic effects slightly enhance binding despite steric constraints. Among the [6]CPPDs complexes, the encapsulation of CNT(4,4) stands out with a notable interaction energy of −41.31 kcal·mol−1, comparable to the stability observed in (5,5)@[6]CPPAs complexes. However, the (5,5)@[6]CPPDs complex shows a positive Gibbs free energy (ΔG = + 21.71 kcal/mol), indicating non-spontaneous binding due to severe steric hindrance.
The encapsulation of fullerenes within [6]CPPAs is primarily stabilized by π-π stacking and van der Waals interactions. The C60@[6]CPPAs complex exhibits moderate binding affinity (ΔG = −19.95 kcal/mol), with interaction strength influenced by guest curvature and host adaptability. Despite its partial immersion, the C70@[6]CPPAs complex exhibits more favorable complexation (ΔG = −25.19 kcal/mol), suggesting that a larger guest surface area enhances host-guest interactions. The results indicate that C70 is more effectively stabilized within [6]CPPAs than C60, likely due to enhanced dispersion interactions and a better geometric fit.
Fullerene complexation within [6]CPPDs presents greater steric hindrance, resulting in less favorable thermodynamics. The C60@[6]CPPDs complex exhibits a highly unfavorable Gibbs free energy (ΔG = + 30.42 kcal/mol), confirming that the size mismatch between the host and guest leads to destabilized interactions. Conversely, C70@[6]CPPDs forms a more stable complex (ΔG = −19.20 kcal/mol), indicating that it is better accommodated within the CPPDs cavity than C60. The partial immersion of C70@[6]CPPDs, as indicated by di−i distances, results in lower interaction energy. Notably, C70 forms a stable “ball in a bowl” complex more effectively than fully immersed CNT(5,5) and C60, a feature attributed partly to CH.π interactions45. The proximity of host and guest centers enhances crucial interactions that stabilize the complex and increase the overall complexation energy in size-selective complexes. The binding becomes more spontaneous as the CNT(5,5) goes further from the ring center. Similarly, the ball-in-a-bowl configuration of C60 has an interaction energy comparable to that of the [6]CPPAs.
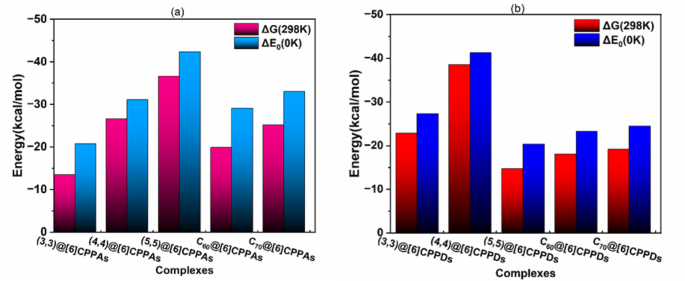
The zero-point corrected binding energy (ΔE0) and change in Gibbs free energy (ΔG298K) plotted for each complex of (a) [6]CPPAs and (b) [6]CPPDs.
The thermodynamic trends observed in these host-guest systems underscore the distinct binding preferences of [6]CPPAs and [6]CPPDs. While [6]CPPAs exhibit a size-dependent increase in stability, favoring larger guests such as CNT(5,5) and fullerenes, [6]CPPDs demonstrate a preference for medium-sized guests like CNT(4,4), with larger guests experiencing steric destabilization. The instability of (5,5)@[6]CPPDs and C60@[6]CPPDs suggests that alternative binding models, such as the hand-lever and ball-in-a-bowl configurations, play a crucial role in stabilizing host-guest interactions when steric limitations are present. Figure 5 depicts geometry-optimized structures of these newly designed complexes, such as the “hand-lever” model of (5,5)@[6]CPPDs and the ball-in-a-bowl structure of C60@[6]CPPDs. These structural models highlight how host-guest interactions manifest, influencing stability and functional properties in supramolecular chemistry.
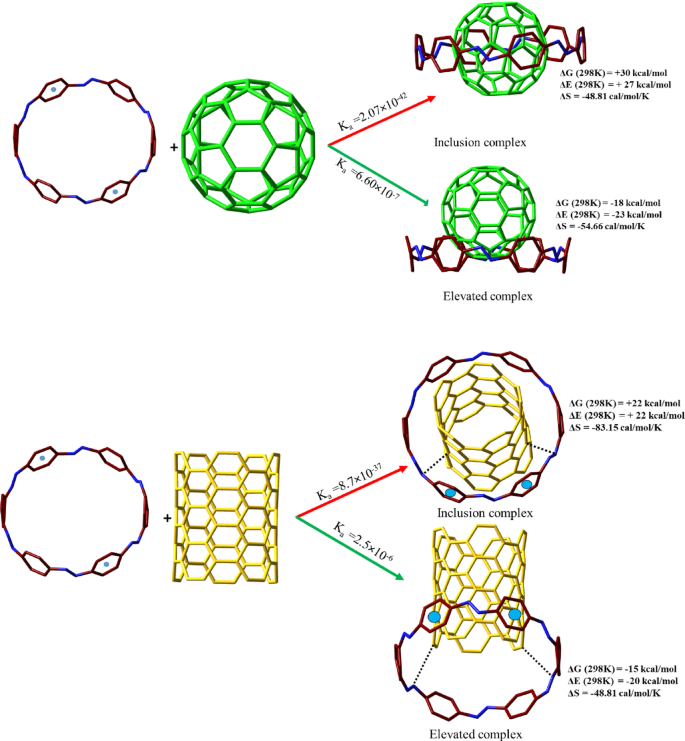
The structural comparison between C60@[6]CPPDs and (5,5)@[6]CPPDs.
By analyzing the binding constants (Ka) at various temperatures, we can infer critical thermodynamic properties, shedding light on the factors influencing the stability and behavior of these molecular complexes. The temperature dependence of the equilibrium constants, as seen in Fig. 6, reveals that the host-guest interactions in both systems are exothermic, as indicated by the increasing Ka values at lower temperatures. This trend aligns with the expected behavior of exothermic processes, where lower thermal energy (lower temperatures) favors stronger binding interactions, leading to higher equilibrium constants. This analysis ties into the established Gibbs free energy, showing that the negative free energy change is directly related to a more spontaneous binding process. A more negative Gibbs free energy signifies a higher equilibrium constant, reinforcing that the interactions are thermodynamically favored as temperature decreases.
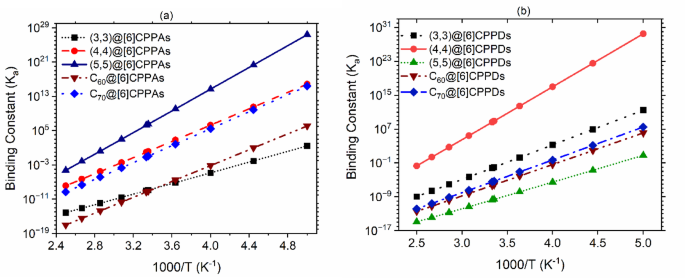
The binding constant (Ka) values for (a) [6]CPPAs complexes and (b) [6]CPPDs complexes. The (5,5)@[6]CPPDs correspond to a hand-lever configuration, whereas the C₆₀@[6]CPPDs exhibit a ball-in-bowl arrangement.
Interaction between complex
Non-covalent forces govern the molecular mechanisms driving these interactions, particularly van der Waals interactions and π-π stacking. These non-covalent interactions are vital in stabilizing the complexes in host-guest systems such as [6]CPPAs or [6]CPPDs rings interacting with fullerenes or CNTs. To better understand thermodynamics and kinetics stability, we analyzed Intermolecular interaction based on scatter plots of the reduced electron density. The hostile regions in the scatter plot correspond to bonding interactions such as hydrogen bonding. In this case, significant van der Waals interactions or π-π stacking occur near the zero region of the plot. The vicinity with a red color or positive value in the scatter plot indicates steric repulsion. The scatter plots for (5,5)@[6]CPPAs, (5,5)@[6]CPPDs, C60@[6]CPPDs, C70@[6]CPPAs, C70@[6]CPPDs are shown in Fig. 7 and for and for (3,3) and (4,4)CNTs, are shown in Supporting information Figure S1 to S6. For the (3,3)@[6]CPPAs complex, van der Waals attraction forces are prominent on the distorted side. The discontinuation or breakage of density can be attributed to the geometrical distortion from the center. The part of the nanotube far from the host experiences no interactive forces, suggesting that van der Waals forces alone are responsible for the observed distortion. Additionally, repulsion between the nanotube and the benzene rings of the host ring is also apparent. Within the CNT, steric repulsion is evident.
In the scatter plot of the [6]CPPDs counterpart, ring-based steric repulsion is prominent, as indicated by the positive values. The denser gradient of van der Waals interaction is closer to the region where the CNT(4,4) and [6]CPPAs rings are nearby, likely causing the complex’s distortion. Here, repulsion within the tube is also observed. There is a slight density decrease in the (4,4)@[6]CPPDs complex where the guest is far from the host, suggesting that the main interacting forces are van der Waals interactions and steric repulsion. Notably, the [6]CPPDs complex exhibits more van der Waals interaction than its [6]CPPAs counterpart.
The RDG analysis of (5,5)@[6]CPPAs and (5,5)@[6]CPPDs reveals distinct non-covalent interaction patterns governing their stability. In (5,5)@[6]CPPAs, van der Waals interactions dominate, with a balanced charge distribution and minimal steric repulsion, leading to more substantial host-guest accommodation. In contrast, (5,5)@[6]CPPDs exhibit stronger steric repulsion and asymmetric charge localization, primarily due to nitrogen-induced electrostatic effects. The scatter plots confirm that (5,5)@[6]CPPDs have more repulsive interactions, leading to weaker binding (ΔG = 21.71 kcal/mol) compared to (5,5)@[6]CPPAs (−36.68 kcal/mol). The increased rigidity of [6]CPPDs limits its ability to encapsulate larger guests like CNT(5,5), whereas [6]CPPA’s flexible framework allows for better host-guest interactions. C60@nanohoop has a uniform distribution of forces that help the guest hold within the host. Except for the repulsion from the benzene rings in [6]CPPAs and the aversion among the carbon atoms in C60, no other significant repulsive forces are acting on this complex. This could be the reason why it is floating over the ring. A powerful steric repulsion has been found in the case of C60@[6]CPPDs. The almost symmetric immersion of C60 in the host might be due to this repulsion; the strong aversion among themselves and the ring might not have allowed the guest to immerse in complete symmetry. Even though Van der Waals’s force is seen in the plot, the repulsive forces dominate. Notice that the ball in a bowl model of C60@[6]CPPDs has more vital π-π concave-convex interaction than the immersed one. Also, the bonding interaction is sharper. This could be the reason for the floating model’s higher complexation energy and stable kinetics. Furthermore, nitrogen substitution in the [6]CPPDs systems introduces additional thermodynamic and electrostatic considerations. Nitrogen in the diazene moieties of [6]CPPDs nanorings creates regions of partial positive charge, which can engage in electrostatic interactions with the fullerene or CNT guests. The “Hand-lever” configuration for CNT (5,5) also benefits from nitrogen-induced polarizability, resulting in higher Keq values and more robust binding as temperature decreases. Nitrogen enhances the host’s ability to stabilize larger guest molecules through inductive effects, increasing the binding strength and favorability of the host-guest interactions49.
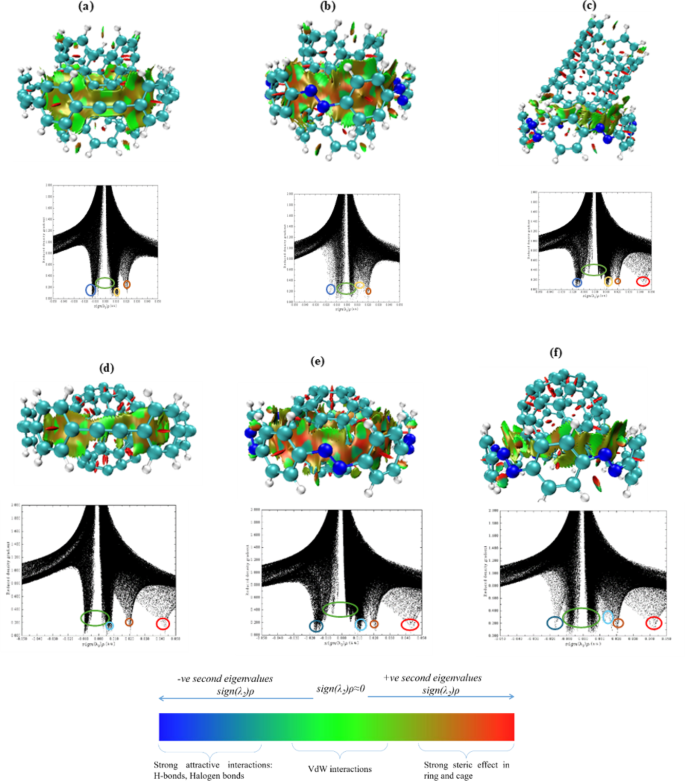
The visualized weak interaction regions and corresponding scatter plots of; (a) (5,5)@[6]CPPAs, (b) (5,5)@[6]CPPDs, (c) Hand-lever model (5,5)@[6]CPPDs, (d) C60@[6]CPPAs, (e) C60@[6]CPPDs, (f) Ball-in-a-bowl C60@[6]CPPDs. The chromatic reference scale is shown in the color diagram.
The π-π interactions also contribute significantly to the binding, as these interactions arise from the overlap of delocalized π-electron systems between the [6]CPPAs or [6]CPPDs host and the guest molecule. This overlap is particularly effective when the guest is a fullerene or CNT with large, conjugated π-systems. The thermodynamic stability of these interactions increases as the temperature decreases, allowing for closer proximity and more pungent π-π stacking between the host and guest molecules50.
The QTAIM molecular graphs of the CNT host-guest complexes offer crucial insights into the nature and extent of non-covalent interactions governing their supramolecular behavior, as shown in Fig. 8(for 5,5, C60 and C70) and Supporting Information Figures S7 and S8 (for 33, 4,4). For the CNT(3,3) encapsulated in [6]CPPAs, a clear pattern of bond critical points (BCPs) emerges, with orange bond paths connecting the carbon atoms of the cyclic host to those of the guest nanotube. These interactions are symmetrically distributed around the nanotube axis, confirming a coaxial alignment and supporting stable π–π stacking interactions between the curved π-systems. As the CNT diameter increases to (4,4), a corresponding increase in the number and distribution of BCPs is observed, suggesting improved spatial complementarity with the [6]CPPAs ring. The bond paths in this complex are more radially distributed, covering the guest’s outer surface more evenly, which indicates a more extensive and uniform interaction zone. This shift strengthens the host-guest binding and aligns with energetic trends that favor CNT(4,4) over CNT(3,3) encapsulation.
This trend culminates in the (5,5)@[6]CPPAs complex, which displays the most extensive and continuous BCPs network among the [6]CPPAs-hosted systems. The bond paths cover both the equatorial and axial regions of the guest, forming a cylindrical shell of interactions that reflects an almost ideal dimensional match between the host cavity and the nanotube. This configuration represents the most stable [6]CPPAs-based complex, confirming that optimal geometric alignment enhances non-covalent stabilization.
Transitioning to the nitrogen-containing [6]CPPDs systems, a comparable pattern is noted in the (3,3)@[6]CPPDs complex. Here, incorporating diazene (N = N) units introduces electronic asymmetry to the host, leading to a skewed distribution of BCPs. These bond paths still prominently connect the host’s inner wall to the nanotube. However, they are slightly denser near nitrogen-rich zones, suggesting localized electrostatic stabilization in addition to π–π interactions. Notably, compared to (3,3)@[6]CPPAs, the [6]CPPDs host enhances directional interactions due to its polarizable π-surface, possibly improving selectivity and responsiveness.
In the case of the (4,4)@[6]CPPDs complex, the QTAIM graph reveals more peripheral and asymmetric BCP distribution, reflecting the looser fit of the slightly larger CNT within the host cavity. Nonetheless, nitrogen atoms once again appear to guide and enhance localized interactions, especially where the curvature matches most closely. The diazene units seemingly compensate for reduced van der Waals overlap by facilitating polarization-induced stabilization. This behavior reflects the improved thermodynamic favorability of this complex over its CPPA counterpart, as seen in the calculated ΔG and binding energy values.
However, the (5,5)@[6]CPPDs complex presents an intriguing divergence. Although its molecular graph shows numerous BCPs connecting the nanotube and the host, indicating significant non-covalent contact, the overall interaction is thermodynamically unfavorable. Both the counterpoise-corrected interaction energy and ΔG are positive, suggesting that geometric strain, repulsion, or curvature mismatch negates the stabilizing effect of the observed electron density pathways. This highlights a key insight: multiple BCPs do not necessarily imply thermodynamic favorability. In this case, the rigidity and polarity introduced by the N = N linkers in the [6]CPPDs host may hinder its ability to conform to more prominent guests, such as the CNT(5,5), despite an apparent topological connection.
This limitation is resolved in a modified configuration, the “hand-lever” (5,5)@[6]CPPDs complex, where the nanotube sits above the ring rather than fully encapsulated. The QTAIM graph here shows an asymmetric but rich network of BCPs, and more importantly, the interaction is thermodynamically favorable. The negative ΔEcp. and ΔG values suggest that this partial immersion strikes an optimal balance between dispersive interactions and steric relief. The flexibility imparted by the diazene linkers allows the host to adapt to this non-standard geometry, leading to a stable, albeit unconventional, host-guest interaction. This adaptation reflects the nuanced role of electronic and geometric tuning in supramolecular assembly. It underscores the potential of nitrogen-doped systems for hosting a broader range of molecular guests through flexible or semi-open configurations.
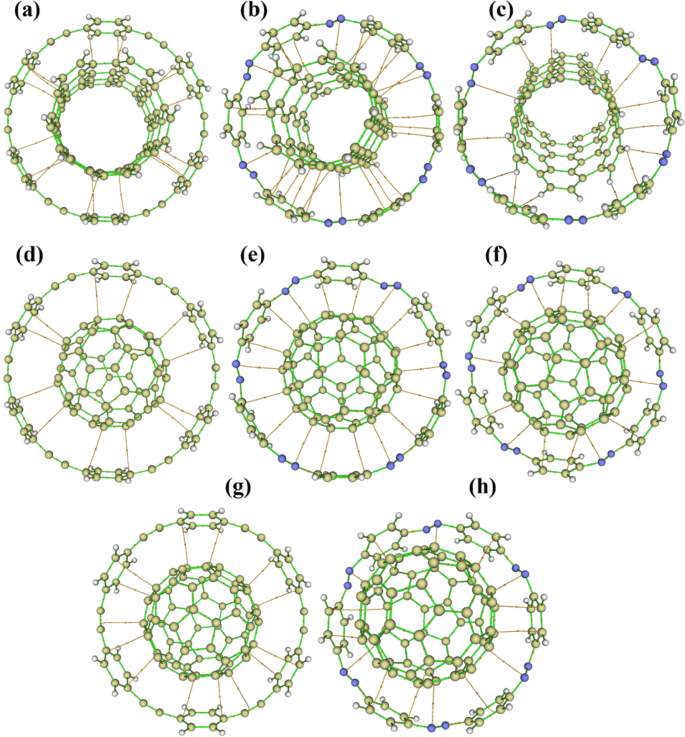
QTAIM molecular graph for; (a) (5,5)@[6]CPPAs, (b) (5,5)@[6]CPPDs, (c) Hand-lever model (5,5)@[6]CPPDs, (d) C60@[6]CPPAs, (e) C60@[6]CPPDs, (f) Ball-in-a-bowl C60@[6]CPPDs, (g) C70@[6]CPPAs, (h) C70@[6]CPPDs. Lines connecting the nuclei are the bond paths. Small orange dots correspond to BCPs.
In the case of C₆₀@CPPAs, the visualization exhibits a highly symmetric cage-like interaction, where the fullerene is centrally positioned within the macrocyclic ring. This central alignment results in evenly distributed BCPs radiating from the surface of C₆₀ to the inner wall of [6]CPPAs. These uniformly spaced bond paths suggest isotropic van der Waals interactions and π–π stacking, stabilizing the encapsulated guest via weak but extensive dispersion forces. The radial symmetry of the BCP network confirms strong encapsulation with minimal directional bias, reflecting optimal shape complementarity between the spherical guest and the cyclic host.
By contrast, the encapsulation of C₇₀ within [6]CPPAs yields a distinctly asymmetric QTAIM signature. The ellipsoidal shape of C₇₀ introduces curvature mismatch, resulting in an uneven distribution of BCPs. These interactions are most concentrated along the “waist” of the C₇₀ molecule, its equatorial region where the surface comes closest to the host ring. Fewer or more elongated bond paths are observed at the poles, underscoring reduced interaction strength due to geometric misalignment. This partial asymmetry aligns with the characteristic floating configuration reported for similar systems, where the fullerene guest sits deeper into the ring without complete immersion. Despite the asymmetry, these interactions remain strong, and the thermochemical data corroborate this, showing favorable binding energies and spontaneous encapsulation for both C₆₀ and C₇₀ in [6]CPPAs.
Transitioning to [6]CPPDs-based hosts, the encapsulation dynamics shift significantly. The molecular graph of C₆₀@CPPDs displays a network of bond paths similar in spatial arrangement to that of C₆₀@[6]CPPAs, indicating the geometric feasibility of encapsulation. However, despite the presence of multiple BCPs, the counterpoise-corrected interaction energy and Gibbs-free energy for this complex are both positive, indicating that the encapsulation of C₆₀ in [6]CPPDs is thermodynamically unfavorable. This disconnect between topology and energetics likely arises from the electronic influence of the diazene (N = N) units in the [6]CPPDs framework. While they introduce dipolar and potentially interactive sites, they also disrupt uniform dispersion interactions or induce steric/electrostatic repulsions that negate the stabilizing effects of π–π stacking. Thus, although electron density bridges exist between host and guest atoms, they do not correspond to energetically viable complexation.
This contrast becomes even more striking when comparing the regular C₆₀@[6]CPPDs complex with its “ball-in-a-bowl” variant, where the fullerene is elevated slightly above the central axis of the ring. In this configuration, QTAIM analysis reveals a directional, bowl-like pattern of BCPs focused predominantly on one hemisphere of the C₆₀, specifically the lower portion interfacing closely with the [6]CPPDs ring. This spatial reorganization reduces steric congestion and maximizes interaction efficiency without inducing substantial strain on the host. The thermodynamic data affirm this, with both ΔEcp. and ΔG being negative, indicating spontaneous and favorable complexation. The elevated position of the fullerene increases the entropy of the system by reducing confinement and facilitates a broader interaction surface. This configuration offers a unique potential for dynamic applications, including stimuli-responsive molecular shuttles or controlled capture–release systems, where partial insertion could aid in reversibility or guest mobility.
The case of C₇₀@[6]CPPDs showcases the most robust and favorable interaction among all fullerene complexes studied. Here, the QTAIM graph exhibits an extensive and dense network of BCPs, mainly clustered along the equatorial region of the C₇₀ guest. This enhanced contact zone matches well with the wider midsection of the ellipsoidal fullerene, reflecting size and shape complementarity. The nitrogen atoms in [6]CPPDs amplify host-guest affinity by increasing local polarizability and fine-tuning the electronic environment to match the fullerene’s π-surface. This synergy results in both a topologically rich interaction profile and highly favorable thermodynamics, as evidenced by the negative ΔEcp and ΔG values for C₇₀@[6]CPPDs. Compared to the C₆₀-based complexes, this system stands out for its superior compatibility and encapsulation efficiency, emphasizing the role of host flexibility and heteroatom doping in optimizing supramolecular architectures.
The influence of guest encapsulation on the aromatic character of [6]CPPAs and [6]CPPDs hosts is clearly delineated through a combined assessment of NICS(0), isotropic, and anisotropic values (see Supporting Information Table S8). In [6]CPPAs-based complexes with carbon nanotubes, the aromatic system demonstrates notable resilience. The (3,3)@[6]CPPAs complex shows an isotropic NICS value of ~ 0.27 ppm and high anisotropy of ~ 37.39 ppm, indicating intact π-delocalization. As guest diameter increases, a progressive rise in isotropic values is observed, 7.55 ppm for CNT(4,4) and 9.78 ppm for CNT(5,5), accompanied by decreasing anisotropy, reflecting growing disruption to the aromatic ring currents. These results highlight moderate electronic perturbation while preserving the characteristic aromaticity of CPPAs. In contrast, the CPPDS host exhibits a stronger response to CNT encapsulation. The NICS(0) values increase sequentially from 6.80 ppm for (3,3)@CPPDS to 10.46 ppm for the fully inserted (5,5)@[6]CPPDs complex, clearly indicating enhanced π–π interaction and severe aromatic disruption. Interestingly, a partial-insertion “hand-lever” conformation of the CNT(5,5) tube results in a near-zero NICS(0) value (0.0075 ppm), underscoring the importance of spatial overlap in influencing electronic delocalization.
A similar yet more pronounced effect is observed in the fullerene series. In [6]CPPAs complexes, both C₆₀ and C₇₀ induce substantial electronic perturbation, with isotropic NICS values of 8.96 and 28.04 ppm, respectively, and low anisotropy values (2.18 and 8.66 ppm), signifying aromaticity loss and weakened ring currents. The [6]CPPDs host responds even more dramatically to central fullerene inclusion: C₆₀@[6]CPPDs exhibits the highest NICS(0) value in the entire series (12.11 ppm), followed by C₇₀@[6]CPPDs (10.07 ppm), affirming strong aromaticity suppression due to deep guest insertion and extensive π-contact. In striking contrast, a “ball-in-a-bowl” conformation of C₆₀ within [6]CPPDs yields a markedly negative NICS(0) of − 5.83 ppm, suggesting preservation or even enhancement of aromatic character due to optimal curvature matching and minimal electronic disturbance. Overall, these trends reveal a clear relationship between guest geometry and host aromaticity: systems with deeper encapsulation and stronger π–π interactions exhibit substantial aromaticity loss, while geometrically favorable, non-invasive conformations retain or amplify aromatic delocalization. The data collectively support the utility of NICS metrics, particularly when combining isotropic and anisotropic values,in capturing the nuanced interplay between structure and electronic behavior in supramolecular complexes.
Electrostatic potential analysis
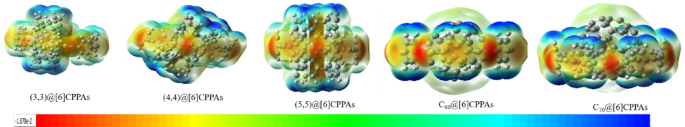
A molecule’s electrostatic potential (ESP) provides deep insights into its interaction energy, especially when considering a unit charge at a specific position in the studied system, excluding charge transfer and polarization effects. The ESP and HOMO-LUMO gap in these host-guest systems reveals a nuanced interplay of electronic stability, photoresponsivity, and structural compatibility23. Additionally, incorporating ESP analysis into current systems enhances understanding of these interactions by shedding light on the interplay between the molecular structures and their surrounding environment, providing a holistic view of the forces governing host-guest complexes. By mapping ESP, we visualized electron density distributions, with red areas representing electron-dense regions and blue areas indicating electron-deficient regions. This color scaling ranges from highly electron-dense regions (red) to moderately dense (yellow/green) and electron-poor areas (blue). These color-coded maps provide a detailed understanding of where electrophilic or nucleophilic interactions are most likely. For instance, in Fig. 9, the ESP diagram of the free guest molecule and free host molecules reveals the regions of high electron density (−0.0295 eV) in red, while the areas of low electron density (+ 0.02953 eV) are in blue. The [6]CPPAs exhibit strong electron localization at the acetylene bridges, creating regions of high negative charge density, which suggests stronger π-π stacking or dipole interactions with guests. In contrast, [6]CPPDs show a more uniform charge distribution, with less intense electron density at the diazene bridges, indicating weaker but more evenly distributed binding interactions. Among the free CNTs, the CNT(3,3) displays strong electron-rich regions, suggesting localized electrostatic interactions but potential strain upon complexation. The CNT(4,4) exhibits a more balanced charge distribution, allowing for stabilized interactions, while the CNT(5,5) has a uniform charge distribution, implying van der Waals-driven interactions rather than electrostatic effects. For the fullerenes, C60 shows minimal charge redistribution, relying on van der Waals interactions, whereas C70 exhibits, more significant electroredistribution, indicating possible charge transfer interactions upon binding.

ESP diagram of the free Guest molecule and free host molecules, the red color corresponds to high electron density (−0.0295 eV), and the blue color corresponds to less electron density (+ 0.02953 eV).
ESP diagram of Comlex@[6]CPPAs. The red color corresponds to high electron density (−0.018 eV), and the blue color corresponds to less electron density (+0.018 eV).
Moving to Figs. 10 and 11, which depict the ESP diagrams of the complex@[6]CPPAs and complex@[6]CPPDs, respectively, we observe similar patterns of electron distribution. In both host systems, the phenyl rings exhibit moderate electron density, while the hydrogen atoms on the rim of the rings serve as centers for electrophilic interactions. The bridges formed by multiple bonds between the phenyl rings are the most electron-rich areas, resulting in an electronegative interior within the rings. However, in nitrogen-containing [6]CPPDs rings, the electron-rich centers are more aligned, suggesting that incorporating electron-deficient moieties into these loops would yield favorable interactions. The nitrogen atoms in the [6]CPPDs rings also enhance the basicity of the phenyl hydrogen atoms compared to [6]CPPAs, further affecting the host-guest interactions.
ESP diagram of comlex@[6]CPPDs; The red color corresponds to high electron density (−0.018 eV), and the blue color corresponds to less electron density (+0.018 eV).
The distortion observed in the geometry of (3,3)@host can be directly attributed to the strong interaction between the electron-rich acetylene bridges and the carbon nanotube guest. The lower atom count in the nanotube results in a stronger attraction, leading to structural strain within the host. Interestingly, the region where the guest is more attracted shows less electron density at the diazene bond than the rest of the host ring. This consistent electron cloud density suggests that the interactions between the host and guest might not involve significant π-π interactions or electron transfer. Instead, these interactions may be driven by weak van der Waals forces or hydrogen bonding. This conclusion could be supported by further analysis of frontier molecular orbitals and reduced density gradient plots.
Similarly, in the (4,4)@[6]CPPDs complex, although the geometry is not as distorted as in the (3,3)@[6]CPPDs complex, the diazene bond’s electron richness is still comparable to the host ring’s. However, the guest’s center in CNT (4,4) has a higher electron density than CNT(3,3), likely due to the more significant number of atoms in the structure. This trend continues in the CNT(5,5) complexes. In the hand-lever model of (5,5)@[6]CPPDs, there is a significant decrease in electron density around the ring and an increase in basicity. The asymmetric leaning of the CNT(5,5) guest over the host ring suggests a shift in the potential surface to achieve stable complexation. This observation aligns with findings from CNT complexes, where the host ring’s electron density remained unchanged except for the elevated guest.
In complexes like C70@[6]CPPDs, the ESP analysis indicates a notable reduction in the potential distribution across the host ring, suggesting the involvement of both host and guest orbitals in complex formation. This could potentially indicate a charge transfer interaction. However, further analysis of the frontier molecular orbitals is necessary to confirm these findings.
The trends observed in electron density variations and potential shifts within host-guest complexes also align well with established interactions in supramolecular chemistry. For instance, the study of size-related π-π overlap phenomena in various ring sizes, especially in C70 complexes, shows that the overlap occurs more prominently as the ring size decreases. The lone pairs from the nitrogen atoms in [6]CPPDs contribute to the ring current, enhancing the photoresponse.
Band gap analysis
In addition to ESP analysis, the HOMO-LUMO gaps of the host-guest complexes provide further insights into these systems’ electronic stability and photoresponsivity. The HOMO-LUMO gap or both complex@[6]CPPAs and complex@[6]CPPDs is shown in Fig. 12, and values are tabulated in Supporting Information Table S9.
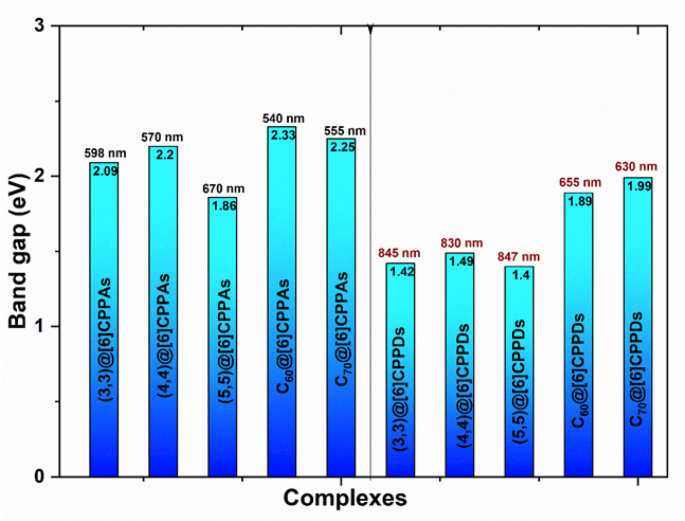
The HOMO-LUMO gaps and λmax of the host-guest complexes.
For the CNT complexes in [6]CPPAs, the (3,3)@[6]CPPAs complex has a band gap of 2.09 eV, indicating a moderate level of electronic delocalization. As the nanotube size increases, the band gap decreases, with (4,4)@[6]CPPAs exhibiting a band gap of 2.2 eV and (5,5)@[6]CPPAs showing the lowest gap of 1.86 eV among the CNT-[6]CPPAs complexes. This trend suggests that larger guest molecules facilitate stronger host-guest interactions, leading to greater orbital overlap and charge delocalization.
A similar trend is observed in CNT complexes in [6]CPPDs but with consistently lower band gaps. The (3,3)@[6]CPPDs complex exhibits a band gap of 1.4 eV, significantly lower than its CPPA counterpart. The (4,4)@[6]CPPDs complex follows with a slightly increased gap of 1.49 eV, while the (5,5)@[6]CPPDs complex also maintains a narrow gap of 1.4 eV. The reduced band gaps across these complexes indicate greater charge mobility and electronic coupling within the [6]CPPDs framework.
For the fullerene complexes in [6]CPPAs, the C60@[6]CPPAs complex has a band gap of 2.33 eV, while the C70@[6]CPPAs complex exhibits a slightly lower band gap of 2.25 eV. This reduction in band gap suggests that C70, with its larger surface area, interacts more effectively with the [6]CPPAs host, leading to greater electronic delocalization. The fullerene complexes in [6]CPPDs follow a similar pattern but with consistently lower band gaps. The C60@[6]CPPDs complex exhibits a band gap of 1.89 eV, while the C70@[6]CPPDs complex has the lowest band gap among the fullerene-[6]CPPDs systems at 1.99 eV. This trend suggests that fullerenes exhibit better electronic coupling with the [6]CPPDs host, further reinforcing the impact of nitrogen incorporation on charge transfer properties. The Frontier Molecular Orbitals are visualized and plotted in Supporting Information Figures S9 to S22.TD-DFT calculations were done to predict the UV-spectrum, and λmax analysis for hosts and host-guest complexes are provided in the Supporting Information (Figures S23 to S27). The λmax values for each complex are shown in Fig. 12.
A clear trend is that [6]CPPDs complexes generally exhibit higher λmax values than their [6]CPPA counterparts, meaning [6]CPPDs absorb light at longer wavelengths. The highest λmax values are observed in (3,3)@[6]CPPDS (845 nm) and (5,5)@[6]CPPDS (847 nm), indicating that steric and electrostatic effects contribute to shifting absorption toward the near-infrared (NIR) region. The higher λmax (718 nm) in the hand-lever model arises from charge delocalization, nitrogen-induced electrostatic effects, and enhanced π-π interactions, making this conformation more electronically active and optically responsive compared to the fully encapsulated (5,5)@[6]CPPDs complex. Similarly, in [6]CPPAs, (3,3)@[6]CPPAs (598 nm) and (5,5)@[6]CPPAs (670 nm) exhibit high λmax values, confirming that size-dependent electronic interactions significantly influence optical transitions. The fullerene complexes (C60@[6]CPPAs, C70@[6]CPPAs, C60@[6]CPPDs, and C70@[6]CPPDs) exhibit relatively consistent λmax values (~ 540–655 nm), suggesting that their electronic transitions are primarily governed by π-π stacking and weak van der Waals interactions rather than significant charge transfer effects. Notably, the ball-in-a-bowl C60@[6]CPPDs complex (636 nm) exhibits a slightly lower λmax than the standard C60@[6]CPPDs (655 nm), reinforcing the hypothesis that binding mode plays a role in modulating electronic transitions.
Size-Selective and photoresponsive characters
In general, in the case of CNT complexes with [6]CPPAs, a size-dependent trend is observed, whereas larger CNT guests exhibit more substantial stabilization. Among the CNT@[6]CPPAs complexes, (5,5)@[6]CPPAs demonstrate the most favorable binding (ΔG = −36.68 kcal/mol, Ka = 3.1 × 10⁶), indicating a highly spontaneous interaction, followed by (4,4)@[6]CPPAs (ΔG = −26.69 kcal/mol, Ka = 1.4), which also exhibit strong thermodynamic preference. In contrast, (3,3)@[6]CPPAs (ΔG = −13.55 kcal/mol, Ka = 1.3 × 10⁻⁹) shows the weakest binding among the CNT complexes, suggesting that smaller CNTs do not fit as effectively within the [6]CPPAs cavity. The trend indicates that encapsulation becomes more efficient as the CNT size increases, leading to more stable host-guest interactions. In the case of CNT encapsulation within [6]CPPDs, a different trend follows, where intermediate-sized CNT guests exhibit the strongest stabilization, particularly (4,4)CNT. The (4,4)@[6]CPPDs complex shows the most favorable binding (ΔG = −38.56 kcal/mol, Ka = 7.8 × 10⁸), suggesting robust and selective binding. Meanwhile, (3,3)@[6]CPPDs (ΔG = −22.92 kcal/mol, Ka = 9.2 × 10⁻³) exhibit moderate stability but are significantly lower than (4,4). A notable deviation is observed in (5,5)@[6]CPPDS, which shows a positive ΔG (21.71 kcal/mol, Ka=8.72 × 10−37), making its encapsulation thermodynamically unfavorable. This instability is likely due to steric hindrance. However, the hand-lever configuration in CNT(5,5) is thermodynamically more stable than the former (ΔG = −14.72 kcal/mol, Ka = 2.45 × 10−10), which is not fully immersed within the [6]CPPDs cavity.
In the case of fullerene@[6]CPPAs complexes, C70@[6]CPPAs (ΔG = −25.19 kcal/mol, Ka =1.1 × 10−1) exhibits stronger binding than C60@[6]CPPAs (ΔG = −19.95 kcal/mol, Ka =1.1 × 10−1), suggesting that C70 fits better within the [6]CPPAs cavity and forms stronger π-π interactions. For fullerene@[6]CPPDs complexes, a striking difference is observed compared to [6]CPPAs. C60@[6]CPPDs exhibits a positive ΔG (30.42 kcal/mol, Ka = 2.07 × 10−42) making its encapsulation thermodynamically unfavorable. This suggests that steric hindrance and electronic factors prevent efficient binding within the rigid CPPDs cavity. The ball-in-a-bowl configuration observed for C60@[6]CPPDs complexes suggests an alternative binding mode, where steric repulsion prevents complete encapsulation, but concave-convex interactions contribute to stabilization (ΔG = −18.10 kcal/mol, Ka = 6.58 × 10−7). In contrast, C70@[6]CPPDs shows a more favorable interaction (ΔG = −19.20 kcal/mol, Ka= 4.94 × 10−06), indicating that C70 is better accommodated within [6]CPPDs than C60. However, it still exhibits weaker stability compared to its [6]CPPAs counterpart. This implies that [6]CPPDs are less efficient than CPPAs in stabilizing fullerene guests, likely due to their electronic distribution and steric rigidity.
The ESP maps of [6]CPPAs host-guest complexes reveal key charge distribution patterns that influence binding strength, stability, and interaction mechanisms. The (3,3)@[6]CPPAs complex exhibits high electron localization at the acetylene bridges, with visible red regions indicating areas of strong electron density accumulation. However, the overall charge distribution remains more localized, suggesting that interactions are limited to specific areas rather than being uniformly spread across the host-guest interface. Moving to (4,4)@[6]CPPAs, the electron density becomes more balanced, with a noticeable reduction in highly localized charge regions, indicating an improved interaction between the host and guest. The (5,5)@[6]CPPAs complex exhibits the most uniform charge distribution, with reduced red regions and broader yellow-green areas, suggesting that the interaction is more delocalized across the entire system. This implies that π-π interactions are maximized in this configuration, leading to a more stable electronic structure.
For the fullerene@[6]CPPAs complexes, the ESP maps reveal significant electron redistribution at the host-guest interface. In C60@[6]CPPAs, the charge density remains relatively uniform, with moderate electron accumulation near the fullerene surface, indicating van der Waals-driven interactions. However, in C70@[6]CPPAs, more pronounced red regions appear along the fullerene-host interface, suggesting more excellent charge localization and stronger electrostatic interactions between the host and guest. This enhanced electron density alignment may facilitate improved orbital overlap, making the interaction stronger compared to C60.
The ESP maps reveal charge distribution variations in [6]CPPDs host-guest complexes, highlighting differences in binding strength, interaction mechanisms, and structural adaptability. In (3,3)@[6]CPPDs, strong electron localization at the acetylene bridges suggests high electrostatic attraction, similar to CPPA, but with less strain due to nitrogen-induced charge alignment. As the guest size increases, (4,4)@[6]CPPDs exhibit balanced electron density distribution, leading to stable host-guest interactions. The electrostatic component weakens in (5,5)@[6]CPPDs, indicating a transition to van der Waals-driven interactions. The hand-lever model of (5,5)@[6]CPPDs further reduces host-guest electrostatic attraction, displaying an asymmetric charge shift to attain thermodynamic feasibility.
C60@[6]CPPDs show minimal charge redistribution for fullerene complexes, confirming that van der Waals forces dominate encapsulation. However, stronger charge localization around C60 suggests better binding affinity due to enhanced electrostatic stabilization in the ball-in-a-bowl model. In C70@[6]CPPDs, significant charge redistribution and reduced electron density near the host ring indicate stronger π-π interactions and possible charge transfer effects, making it the most electronically responsive complex.
For (3,3)@[6]CPPAs, van der Waals forces dominate, particularly on the distorted side, while steric repulsion between the CNT and benzene rings prevents complete encapsulation. The (4,4)@[6]CPPDs complex exhibits stronger van der Waals interactions than its CPPAs counterpart, but steric repulsion within the ring leads to structural distortion. In (5,5)@[6]CPPAs, van der Waals interactions and minimal steric repulsion enable effective host-guest accommodation, supported by its flexible framework and firm binding. In contrast, (5,5)@[6]CPPDs exhibit higher steric repulsion and asymmetric charge localization due to nitrogen-induced electrostatic effects, leading to weaker binding. The hand-lever model in [6]CPPDs benefits from nitrogen-induced polarizability, enhancing electrostatic interactions and improving host-guest stabilization, making it a viable alternative for CNT encapsulation.
In fullerene complexes, C60@[6]CPPAs show a uniform distribution of attractive forces, ensuring stable encapsulation with minimal repulsion. In contrast, C60@[6]CPPDs exhibits strong steric repulsion, preventing symmetric immersion. The ball-in-a-bowl model of C60@[6]CPPDs enhances π-π concave-convex interactions, leading to higher complexation energy and improved kinetic stability. Overall, [6]CPPDs exhibit stronger van der Waals interactions but greater steric repulsion than [6]CPPAs, affecting host-guest accommodation and stability. The findings reinforce how electronic effects, steric hindrance, and weak interactions drive supramolecular behavior in these systems.
A key observation found via HOMO-LUMO analysis is that [6]CPPDs generally exhibit lower band gaps (1.4–1.99 eV) compared to [6]CPPAs (1.86–2.33 eV), suggesting that [6]CPPDs are more electronically active and better suited for optoelectronic applications. Among the [6]CPPDs complexes, (3,3)@[6]CPPDs and (5,5)@[6]CPPDs exhibit the lowest band gaps (1.4 eV), indicating high electronic activity but reduced thermodynamic stability due to steric repulsion and weaker host-guest interactions. In [6]CPPAs, (5,5)@[6]CPPAs have the lowest band gap (1.86 eV), confirming that larger guests induce stronger host-guest interactions, leading to better electronic coupling. Additionally, the hand-lever model of (5,5)@[6]CPPDs exhibits an even lower band gap of 1.35 eV, suggesting that this alternative binding configuration enhances charge delocalization, potentially altering electronic properties in a way that could influence device applications. Meanwhile, the ball-in-a-bowl model of C60@[6]CPPDs has a slightly higher band gap (1.95 eV) compared to the standard C60@[6]CPPDs (1.89 eV), reinforcing the idea that concave-convex interactions enhance charge stabilization, affecting the electronic structure.
While [6]CPPAs primarily stabilize guests based on size compatibility, [6]CPPDs exhibit a greater influence from charge redistribution and host polarity, affecting the complexes’ overall stability and electronic behavior. These findings establish a fundamental contrast between the two host systems, with [6]CPPAs favoring geometric fit and [6]CPPDs displaying significant charge-driven interactions, influencing host-guest complexation trends. The comparison among these host-guest complexes is summarised and tabulated in Supporting Information Table S10.
The contrasting host–guest behaviors observed in [6]CPPAs and [6]CPPDs highlight their potential as functional materials in advanced supramolecular systems. The enhanced electronic activity of [6]CPPDs, particularly in configurations such as the hand-lever and ball-in-a-bowl complexes, accompanied by their reduced band gaps and localized charge redistribution, points to their utility in photoresponsive encapsulation–release systems and stimuli-controlled molecular switches, key components in molecular machines and innovative delivery platforms51,52. The significant host-guest interaction anisotropy and tunable π–π interactions in nitrogen-rich CPPDs also pave the way for their integration into organic optoelectronic devices, including organic photovoltaics and molecular semiconductors, where their curvature and electronic modulation play crucial roles in charge transport and exciton dynamics53. Recent studies have demonstrated the potential of nitrogen-doped cycloparaphenylenes for CO₂ capture through tunable non-covalent interactions, reporting significant complexation energies and spectroscopic responsiveness54. In this context, our comparative investigation of [6]CPPAs and [6]CPPDs with fullerene and carbon nanotube guests offers compelling insights into how macrocyclic architecture and electronic modulation via diazene linkages can influence binding affinity, electron distribution, and interaction topology. The enhanced interaction anisotropy and polarizability observed in [6]CPPDs suggest that these systems could be extended to applications in selective gas adsorption or molecular sensing, where curvature, charge distribution, and responsive encapsulation are critical. The integration of energetic, topological, and electronic descriptors in this study demonstrates that [6]CPPDs, alongside their CPPAs counterparts, can be strategically developed as multifunctional platforms for applications in molecular electronics, sensing, and supramolecular nanotechnology. The broader implications of this research point to [6]CPPDs as a promising class of host molecules with the potential for various applications, such as molecular sensing, drug delivery, and optoelectronic devices. Findings from this study contribute to a deeper understanding of supramolecular chemistry and open avenues for developing advanced materials tailored for specific applications.