Nintendo Switch 2 just got its indie power-up – and it is wild: a Zelda-like game with a roguelike bite, a psychedelic Souls-like co-op, and Super Meat Boy going full 3D chaos.
At every Game Developers Conference, Nintendo highlights projects…

Nintendo Switch 2 just got its indie power-up – and it is wild: a Zelda-like game with a roguelike bite, a psychedelic Souls-like co-op, and Super Meat Boy going full 3D chaos.
At every Game Developers Conference, Nintendo highlights projects…

Alongside cognitive impairment and urinary incontinence, gait disturbance is a cardinal symptom of normal pressure hydrocephalus (NPH). People with NPH can experience slower movements, gait freezing and trouble lifting their feet, which may be caused by irregular cerebrospinal fluid (CFS) distribution and dynamics.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
In a recent trial involving 20 individuals with NPH, magnetic resonance fingerprinting (MRF), a fast quantitative imaging technique, accurately predicted which participants would experience gait improvements after extended lumbar cerebrospinal fluid drainage (ELD).
Cleveland Clinic researchers published results of their prospective cohort study in the Journal of Magnetic Resonance Imaging.
The goal of the study was to assess whether MRF – a non-invasive and relatively quick procedure – could reliably predict which patients with NPH would and would not respond to permanently placed shunt catheters.
Results showed that MRF T1 and T2 relaxation-time measures were significantly associated with gait assessment metrics. If those results are sustained in larger studies, patients with NPH might someday be able to avoid more invasive preoperative assessments, such as lumbar puncture testing, to determine the likely success of shunt surgery.
“The holy grail might be that one day we don’t have to do any (invasive) testing,” says Cleveland Clinic neurologist and corresponding author Stephen E. Jones, MD, PhD. “If Mrs. Smith has a gait disorder, rather than having a two-day inpatient stay, we might be able to do the fingerprinting exam and the mathematical analysis and say the data indicates she will or will not benefit from having a ventriculostomy catheter implanted.”
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/c0da0723-730a-45e5-b576-45d5cc21aca2/mri-images-for-hydrocephalus-study-inset)
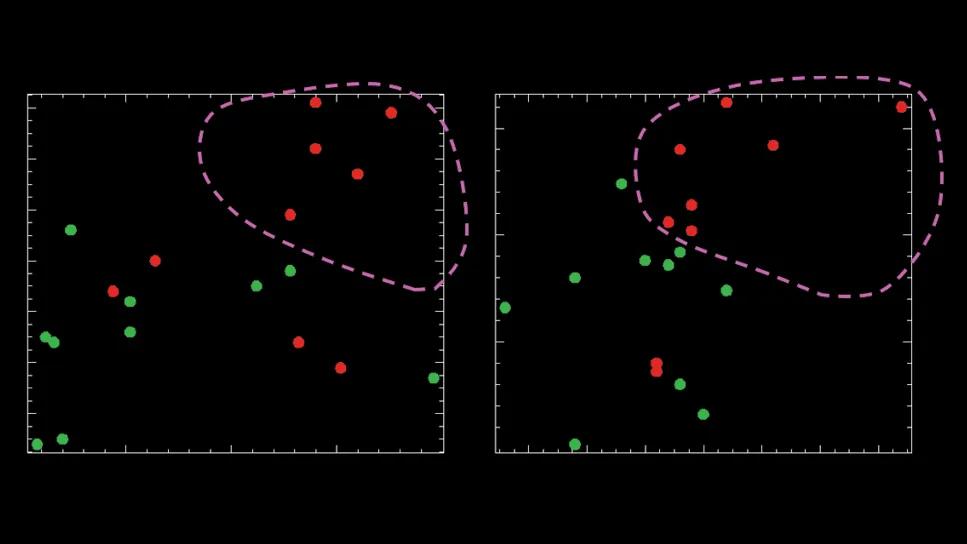
Two-dimensional scatter plot of peak T1 versus T2 times from pre-extended lumbar drainage MRI, for white matter (left) and gray matter (right).
Participants were adults with presumed NPH (idiopathic or secondary), progressive gait disorders, and urinary incontinence or cognitive dysfunction. They were recruited from a group of patients who had been referred for external lumbar drainage trial. A movement disorders neurologist, neuropsychologist and neurosurgeon screened candidates for comorbidities that could confound study results.
Participants were hospitalized for two nights and three days. They underwent initial MRF and clinical assessments, then performed multiple tests, including a 10-meter walking test, during which the number of steps and the time taken were recorded.
The lumbar drain was then inserted, and a standard 36-hour ELD trial began. Ten milliliters of CSF were removed every hour for 36 hours. After completion of ELD, the walking test, MRF and clinical assessments were repeated.
Participants were considered treatment-responsive if the number of steps needed to walk 10 meters was reduced or stayed the same; the time to complete the walk was reduced by at least one second; and a neurosurgeon noted overall improvement in the participant’s condition.
The study used a 3T MRI scanner and implemented a 2D MRF sequence that captured 20 axial slices of 5 mm thickness at 19.5 seconds per slice.
To generate maps of T1 and T2 relaxation times, the signal evolution at each voxel matched against a database of signal patterns, known as the “MRF dictionary.”
Brain metrics before and after drainage were analyzed to determine whether the procedure caused meaningful changes in relaxation times. The study also compared relaxation times in study participants to normal values from previously gathered healthy controls, and evaluated differences in demographics between responders and non-responders.
After ELD, T1 relaxation times showed no consistent changes for white matter, but for gray matter there was a noticeable difference. Responders tended to have lower T1 times after treatment, while nonresponders often had higher T1 times. This suggests that changes in T1 times for gray matter may be linked to treatment effectiveness.
Most participants showed reduced T2 relaxation times in both GM and WM after treatment regardless of whether they experienced clinical improvement. Higher pre-treatment peak T1 and T2 relaxation times in GM or WM were associated with less post-treatment gait improvement.
Developed at Case Western Reserve University by a team led by physicist Mark Griswold, PhD, a coauthor on the study, MRF earned FDA approval for clinical use in 2023. Cleveland Clinic has applied it in epilepsy clinical trials and continues to explore its utility.
Beyond access to the MRF technology itself, says Dr. Jones, clinical application requires advanced analytic tools.
“Almost anyone could do this, but they would have to follow our analysis method. We had to get the pictures and then put them onto a laboratory computer, and I wrote the program that analyzes them,” he says. “We also need a good high-resolution image of brain structure volumetrics. That is going to be something that has like one-millimeter isotropic resolution, so it looks at the brain with very fine detail.”
The researchers hope to continue the inquiry with a larger cohort and an expanded timeline that considers durability of shunt performance.
“We would like to do fingerprinting a year later and see how the participants’ brains changed with the shunt,” says Dr. Jones. “Could we predict instances where too much damage has already been done and the shunt might fail or might help for only a short time? Can we learn anything about those who continue to benefit and those who do not?”
The AI-CS framework offers a promising alternative for cardiac MRI examinations in the clinical setting, where long acquisition time remains a major challenge
Nan Zhang
“For patients with severe arrhythmias, breath-holding can be particularly…


The Khyber Pakhtunkhwa Local Government Department has announced 100 per cent work-from-home policy on Fridays, under austerity drive amid fuel crises after US-Israel and Iran war, ARY News reported.
A formal notification has been issued by the…


Lahore Qalandars began their title defense in the 2026 Pakistan Super League (PSL 11) with a commanding 69-run victory over debutants Hyderabad Kingsmen at Gaddafi Stadium.
Chasing a challenging target of 200 in their first-ever PSL…