
Police stand guard outside the US consulate in Karachi. Photo: file
KARACHI:
…

March 5 (UPI) — NASA on Thursday walked back a prediction that an asteroid had a “small, but notable” chance of impacting Earth or the moon in 2032 based on newly analyzed data.
Scientists said that near-Earth asteroid 2024 YR4 is expected to…

As a child in Newark, N.J., Narciso Rodriguez was often transported back to Cuba by the stories from his family and their friends. He walked the halls of El Encanto, a Havana department store and fashion mecca on the island — one that drew in…
CALGARY, AB, March 5, 2026 /PRNewswire/ – Frontera Energy Corporation (TSX: FEC) (“Frontera” or the “Company“) announces today that the Frontera Board of Directors, in consultation with its external legal counsel and independent financial advisors, has determined that the binding offer (the “Parex Offer“) received from Parex Resources Inc. (“Parex“) to acquire all of Frontera’s upstream Colombian exploration and production business (the “Frontera E&P Assets“) constitutes a “Superior Proposal” (as defined in the GeoPark Arrangement Agreement described below).
Under the Parex Offer, Parex would acquire the same assets that Frontera has agreed to sell to a subsidiary of GeoPark Limited (“GeoPark“) under the previously announced arrangement agreement between Frontera and GeoPark dated January 29, 2026 (the “GeoPark Arrangement Agreement“). The purchase price under the Parex Offer consists of (a) US$500,000,000 in cash payable upon closing; plus, as is the case with the GeoPark transaction (b) an additional US$25,000,000 contingent payment payable upon the achievement of specified development milestones within a period of up to 12 months following the transaction closing date, and (c) the assumption of all of Frontera’s obligations under the US$310,000,000 aggregate principal amount of outstanding 2028 Frontera Unsecured Notes and the US$80,000,000 outstanding under Frontera’s previously announced prepayment facility with Chevron Products Company. The consideration offered in the Parex Offer also assumes the payment of US$25,000,000 Purchaser Break Fee payable to GeoPark by Frontera should Frontera terminate the GeoPark Arrangement Agreement. Except for the consideration being offered, the arrangement agreement that would be entered into with Parex is substantially the same as the GeoPark Arrangement Agreement, and the transaction structure is the same as for the GeoPark transaction.
Frontera has advised GeoPark of this determination, and the five Business Day period (the “Matching Period“) in which, GeoPark has the right, but not the obligation, to amend the terms of the GeoPark Arrangement Agreement in order for the Parex Offer to cease to be a Superior Proposal (the “Match Right“) has commenced. The Matching Period will expire at 11:59 p.m. (Eastern time) on March 12, 2026.
At this time, there can be no assurance that the Parex Offer will result in a transaction or that any transaction contemplated thereby will be completed. The GeoPark Arrangement Agreement remains in effect, and the Frontera Board of Directors continues to act in accordance with its fiduciary duties and the terms of the GeoPark Arrangement Agreement. The Frontera Board of Directors has not changed its recommendation regarding the transaction with GeoPark pursuant to the GeoPark Arrangement Agreement. Frontera will provide updates, including with respect to the determination by GeoPark as to whether or not to exercise its Match Right, as required under applicable securities laws.
About Frontera:
Frontera Energy Corporation is a Canadian public company involved in the exploration, development, production, transportation, storage and sale of oil and natural gas in South America, including related investments in both upstream and midstream facilities. The Company has a diversified portfolio of assets with interests in 18 exploration and production blocks in Colombia and Guyana, and pipeline and port facilities in Colombia. Frontera is committed to conducting business safely and in a socially, environmentally and ethically responsible manner.
If you would like to receive News Releases via e-mail as soon as they are published, please subscribe here: http://fronteraenergy.mediaroom.com/subscribe.
Social Media
Follow Frontera Energy social media channels at the following links:
Twitter: https://twitter.com/fronteraenergy?lang=en
Facebook: https://es-la.facebook.com/FronteraEnergy/
LinkedIn: https://co.linkedin.com/company/frontera-energy-corp.
Cautionary Note Concerning Forward-Looking Statements
This news release contains forward-looking statements. All statements, other than statements of historical facts, that address activities, events or developments that the Company believes, expects or anticipates will or may occur in the future are forward-looking statements. The use of any of the words “estimate”, “will”, “would”, “believe”, “plan”, “expected”, “potential”, and similar expressions are intended to identify forward-looking statements. Forward-looking statements are often, but not always, identified by such words. These statements involve known and unknown risks, uncertainties and other factors that may cause actual results or events to differ materially from those anticipated in such forward-looking statements.
In particular, and without limiting the foregoing, this news release contains forward looking statements with respect to a potential transaction involving Parex and Frontera and the transaction involving Frontera and GeoPark, and the process and timing for both transactions. These forward-looking statements reflect the current expectations or beliefs of the Company based on information currently available to the Company. Forward-looking statements are subject to a number of risks and uncertainties that may cause the actual results of the Company to differ materially from those discussed in the forward-looking statements, and even if such actual results are realized or substantially realized, there can be no assurance that they will have the expected consequences to, or effects on, the Company. Factors that could cause actual results or events to differ materially from current expectations include, among other things: there can be no assurance that any transaction will result from the Parex Offer or that Parex and Frontera will ultimately enter into a definitive agreement for Parex to acquire the Frontera E&P Assets; that the GeoPark transaction will be completed on the terms or within the timeframes currently contemplated; and the failure to obtain all necessary court, third-party and shareholder approvals to complete either such transaction and the risk that either such transaction may be varied, accelerated or terminated in certain circumstances.
Any forward-looking statement speaks only as of the date on which it is made and, except as may be required by applicable securities laws, the Company disclaims any intent or obligation to update any forward-looking statement, whether as a result of new information, future events or results or otherwise. Although the Company believes that the assumptions inherent in the forward-looking statements are reasonable, forward-looking statements are not guarantees of future performance and accordingly undue reliance should not be put on such statements due to the inherent uncertainty therein.
www.fronteraenergy.ca
SOURCE Frontera Energy Corporation


DULUTH, Ga.- The No.7 seed Syracuse women’s basketball team (23-7, 12-6) opened up postseason play in the second round of the 2026 Ally ACC Women’s Basketball Tournament by taking down the No.10 seed California Golden Bears (19-14, 9-9) with a…

New Delhi, India — Dressed in a blue Navy uniform and sleek sunglasses, Indian Prime Minister Narendra Modi, in late October, addressed a gathering of the country’s sea warriors.
He listed out the strategic significance of the Indian Ocean —…

The eighth annual Kodak Film Awards were held Monday evening at the ASC Clubhouse in Hollywood, in a celebration of all things celluloid.
The event’s jubilant tone required a brief reflection on a time when the outlook was not so sunny. Kodak CEO…

The US treasury issued a 30-day waiver on Thursday allowing India to buy Russian oil currently stuck at sea.
“To enable oil to keep flowing into the global market, the treasury department is issuing a temporary 30-day waiver to allow Indian…

Araghchi says ready and ‘waiting’ for US invasion; Tehran strike sparks Bahrain refinery blaze
Smoke rises above the city skyline in Riyadh, amid the US-Israeli conflict with Iran. Photo: Reuters
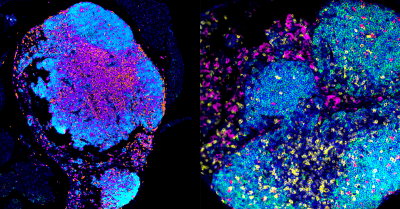
Newswise — Researchers at the University of California San Diego and collaborators have discovered a new way to help the immune system fight ovarian cancer by changing how tumors communicate with nearby immune cells. The…