- Pakistan: harsh winter conditions deepen the humanitarian crisis for Afghan refugees amid forced deportations MSF – Médecins Sans Frontières
- Nearly 350 Afghans released from Pakistani prisons and returned home امید رادیو
- By End of…
Author: admin
-
Pakistan: harsh winter conditions deepen the humanitarian crisis for Afghan refugees amid forced deportations – MSF – Médecins Sans Frontières
-
Dar expresses satisfaction over stock position of sugar – RADIO PAKISTAN
- Dar expresses satisfaction over stock position of sugar RADIO PAKISTAN
- Sugar recovery improves as temperatures fall The Express Tribune
- Prices of essential items show mixed trend Business Recorder
- Traders concerned as govt fixes rates of rice,…
Continue Reading
-

NeurologyLive® Year in Review 2025: What Trials Could Reshape Alzheimer Care? | NeurologyLive
In 2025, the NeurologyLive® staff was a busy bunch, covering clinical news and data readouts from around the world across a number of key neurology subspecialty areas. From major study publications and FDA decisions to societal conference…
Continue Reading
-
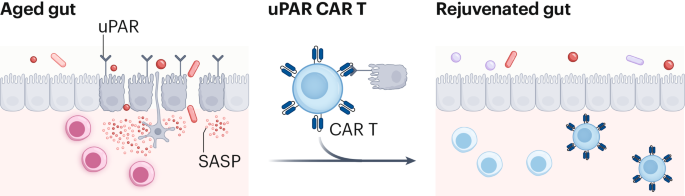
Rejuvenating the aging gut by targeting senescence
Eskiocak, O. et al. Nat. Aging https://doi.org/10.1038/s43587-025-01022-w (2025).
Google Scholar
Delgado-Pulido, S., Yousefzadeh, M. J. & Mittelbrunn, M. Nat. Aging 5, 1393–1403…
Continue Reading
-
Ambreen Jan made new PEMRA chief – Daily Pakistan
- Ambreen Jan made new PEMRA chief Daily Pakistan
- Ex-information secretary Ambreen Jan selected to head Pemra Dawn
- Parliamentary panel shortlists two names for PEMRA chairmanship The Nation (Pakistan )
- Ambreen Jan to become new PEMRA chairman
Continue Reading
-

Hidradenitis Suppurativa Risk Factors Identified
A LARGE two-sample Mendelian randomisation (MR) study provides strong genetic evidence that increased body mass index (BMI) causally increases the risk of hidradenitis suppurativa (HS), while the role of smoking remains unclear. The study also…
Continue Reading
-

911 fails when power is out in Boulder County mountain towns
The drive into Boulder County along Colorado 93 in the winter sometimes has a cinematic quality. The Flatirons are draped in snow, clouds weave in and out of trees, and brisk morning air quickly softens in the midday sun.
But for people who live farther to the west, the mountainous charm can quickly give way to conditions that are unforgiving, shifting from routine to emergency within minutes. Communities behind the Flatirons, like Gold Hill, find themselves at the mercy of severe weather and failing emergency communication systems.
This fragility was on display last month, when hurricane-force winds battered western Boulder County. From midmorning on Dec. 17 through late Dec. 21, the power had been shut off by Xcel Energy in response to warnings of critical fire danger. That risk quickly shifted from precaution to reality: When power was restored, two fires ignited in the area, prompting the need for emergency response. While the fires were extinguished quickly, it was another scary moment for people living in an area increasingly vulnerable to extreme weather.
Worried about the incoming wind storm, disaster managers in Boulder County had asked Lumen Technologies, which provides 911 access in the western half of the county, to prepare for what was forecast and replace the battery backup systems that keep phone service running during power outages — but the company committed no resources.
For Gold Hill, this was not a new problem.
Help was not on the way
Late one night last winter, after a four-day snowstorm, Michael Wollard trudged out of his house in Gold Hill to check on his car. As he pushed through chest-deep snow, cold seeping through the seams of his clothing, he heard a faint meowing — a cat, he thought to himself, was stuck in the street.
The noise quickly shifted from a cat’s meow to a clear human cry: “Help.” In the middle of Main Street, he saw his 85-year-old neighbor trying to navigate the deep snow. The phones were out and her husband was critically ill, so she was left to shuffle through the streets at midnight, relying on her voice to call for help.
At the time, the power was out in Gold Hill. An outdated battery backup system that is supposed to keep residents connected during events like this had failed. Gold Hill was buried, frozen, and cut off from the outside world.
The Finn family are legacy residents of Gold Hill and were volunteer firefighters.
Wollard rushed to the Finn’s house to get help for their neighbors. Unable to move the fire trucks, Leslie Finn and her husband brought out a truck they keep tire chains on at all times. They put the man, who was bleeding internally, in the back of the truck and crept toward Sunshine Canyon, where an ambulance was waiting to take him to the hospital in Boulder.
“It was until 3 a.m. dealing with the craziness,” Finn said. “The whole thing started because we couldn’t call 911.”
In cases of internal bleeding, minutes can mean the difference between life and death. Medical care in these situations is critical, with severe cases leading to death within 5 to 30 minutes on average. The risks are even higher for people in rural communities because of the distance to hospitals.
A home in Gold Hill after a major storm. (Photo courtesy of Rich Caudill) Residents of Gold Hill are already at a disadvantage in emergency situations — without reliable internet service to begin with, inclement weather such as high winds, fires or floods that result in power outages leave Gold Hill cut off from calling for help or receiving emergency notifications, such as orders to evacuate.
In the event of a power outage, the batteries are supposed to kick on, keeping Gold Hill connected for about two hours. Connecting to cellular service is nearly impossible in the town, so landlines and Wi-Fi are the only option. But the batteries have not been replaced for more than 20 years, meaning that they have significantly reduced capacity, according to Gold Hill Fire Protection District Chief Rich Caudill. In the event of a power outage, they fail to keep the communication networks up for more than a few seconds. Gold Hill has no way to contact emergency responders in times of crisis. The blame falls on Lumen, Caudill says.
Lumen Technologies is the sole telecommunications provider in western Boulder County. Formerly known as CenturyLink, the company bought Level 3 Communications in 2017, expanding its network and shifting its focus to wholesale and commercial consumers. Shortly after, the company rebranded as Lumen.
In cars, phones and computers, a two-decade old battery would warrant replacement.
The lifecycle of lithium-ion batteries, like the ones in Gold Hill, has been top of mind in recent years, primarily because discussions of range decrease in electric vehicles. Studies show that modern, highly efficient batteries will decline by 18% by 450 cycles.
While scholarship on older batteries is limited, Gold Hill has proved in nonlaboratory conditions, and over the course of thousands of battery cycles, that Lumen’s batteries are mostly useless holding a connection for maybe 20 seconds after the power goes out, Finn said.

Lumen’s backup battery box is at the eastern edge of Gold Hill. Only authorized employees can access the interior. (Sophia Collins, CU News Corps) Caudill has been taking Lumen to task for years, urging them to upgrade the batteries, the mounting frustrations of his neighbors and friends fueling his fight to hold the corporation accountable for providing crucial service.
“Over the last six months, I have tried reaching them over 15 separate times, with almost no response,” Caudill said in October.
“The town’s interactions with Lumen are minimal,” Caudill said, in most cases met with lukewarm responses like, “We are not able to commit resources at this time,” or are ignored altogether. When the town requests access to the infrastructure to install new batteries using its own tax dollars, that access is denied, he said.
Lumen declined to comment for this story.
However, in documents including communications between Lumen reps and stakeholders, the company has said it would not approve requests for access claiming that using outside equipment or generators could present safety risks. The battery boxes are built only for Lumen’s use, and union workers are the only personnel authorized to handle them.
“They don’t think about the population of Gold Hill … I don’t know … deserves it. This community is not just 100 people, it’s several hundred people, and we all deserve access to 911,” said Maggie Simms, lifelong Gold Hill resident.
Most of Gold Hill’s residents are 60 and older, including most members of the volunteer fire department. There are at least seven original founding families still in Gold Hill, according to Boyd Brown, volunteer historian for the Gold Hill Museum and a sixth-generation Gold Hill resident.
“Residents feel responsible for maintaining the town’s history and mining identity and thus want to stay in the homes that have often belonged to their families for generations,” Brown said. Despite that, they want to be able to rely on the systems in place without putting themselves in danger.

Boulder Valley School District provides the Gold Hill School house with a generator. During power outages, residents can be seen parked outside the building to use the school’s internet. (Sophia Collins, CU News Corps) Residents agree that the situation is dangerous, but many are torn on what to do about it. Starlink, the satellite communications system, presents a reasonable alternative, but one that many residents struggle with because of Elon Musk’s work for the Trump administration. Some see it as a necessity in order to remain connected in a potentially dangerous situation, others are unwilling to make the political compromise.
Starlink may end up being the broadband service Gold Hill and other rural Colorado communities get.
In 2021, Colorado was awarded $826.5 million by the Broadband Equity, Access and Deployment Program in the federal infrastructure bill to help make sure every unserved or underserved household in the state has access to fast internet. This fall, the that sum was cut in half and the state was forced to reapply for funding intended to reach more than 96,000 residents in rural areas. Most of the funding was awarded to satellite broadband proposals, including Starlink and the still unlaunched Amazon Leo, previously known as Project Kuiper.
Most Gold Hill residents don’t expect excellent internet service. Being a bit disconnected, they say, is part of the charm of mountain living, “Our needs are few and simple,” longtime resident Robert Mason said, “Certainly runs everything we need to run, video and blah blah blah.” Mason’s daughter Rebecca Gretz confirmed the feeling, saying that they never had any major problems with their internet.
Emergency communication service is a different matter and it’s hard to argue that the failing battery backups are not an enormous risk to Gold Hill and several other rural mountain communities in Boulder County.
“We’re fighting the law of averages,” said Caudill, the Gold Hill fire chief.

It’s not just a Gold Hill problem
Officials in Gold Hill and neighboring towns are losing patience. The mayor of Jamestown has signed on to partner with Gold Hill’s fire department to pressure Lumen to resolve the battery problem.
Jamestown faces almost identical issues during extreme weather events. In 2013, a flood ripped through the valley, destroying parks, town buildings, including the fire department and 10% of homes in the community. The damage not only prevented residents from physically leaving, but also from getting access to emergency services.
There are day-to-day threats as well. Planned power outages for maintenance or as a precaution during high winds, are common in the region.
In the Fourmile Canyon community, where one of the worst wildfires on record occurred in 2010, Lefthand Fire Protection District Chief Chris O’Brien has been working with county commissioner Steve Silbermann to pressure Lumen to replace the failing backup batteries in all of western Boulder County.
“Although we are making a valiant effort to convince CenturyLink to replace their battery backup power, we don’t have any regulatory backing,” Silbermann said, referring to the company by its former name.
The Boulder County Sheriff’s Office and Boulder County Communications work closely with Lumen, the state-designated Basic Emergency Service Provider, in order to improve 911 reliability. After multiple incidents and discussions, the county requested permission for public safety staff to deploy generators, detailed remote-terminal mapping, critical battery replacements and a battery maintenance plan. Lumen approved only the mapping and battery replacements in crisis situations, according to documents obtained by CU News Corps.
O’Brien said Lumen “has a monopoly” on western Boulder County. “Once they were deregulated it gave them the ability to act with impunity.”
The deregulation O’Brien is referencing is a set of five Colorado laws approved in 2014 that intended to encourage higher broadband internet speeds. At the time of introduction, the bills received criticism from the AARP, who said that the deregulation would hurt landline users. In 2025, it has.
The acquisition of Level 3 Communications that expanded CenturyLink also made Lumen slower to respond, O’Brien said. “When it was CenturyLink, conditions were better … the service technicians really knew the area. It’s hard for us, because we don’t have that, we don’t have a head technician that we can reach out to in case of emergency.”
Allenspark and Raymond on the northern edge of Boulder County have also confirmed that the area is facing similar problems because of Lumen’s failing backups.

Jamestown Fire Chief Seth Strickland warns of the risks that come with losing communication. (Sydney Schrader, CU News Corps) “It’s life or death. If we have a retiree, or even a healthy young person, take a fall, have a cardiac event … and they’re not able to reach 911 for emergency services, we don’t know that they need help,” Jamestown Fire Chief Seth Strickland said.
Until the infrastructure improves, residents are left to their own devices. When outages occur in Gold Hill, this often means driving to the far eastern corner of town for spotty cellphone coverage at best. In some cases, the town congregates around the schoolhouse to use its backup resources — it’s one of the only buildings in town with a built-in generator. The town relies upon methods like this to support itself. In theory, they shouldn’t have to.
“What we’re looking for is safety, we want the best standard of living for our town residents, we are not going to stop working on it until we have the issue solved,” Jamestown Mayor Michael Box said.
Lumen is expected to sell the entirety of its mass-market business to AT&T this year. This means that customers in Boulder County mountain towns may have a fresh chance at connectivity under new management. But Gold Hill and the rest of western Boulder County want the systems already in place must be maintained.
“If I were able to snap my fingers today? I would love to have those batteries replaced and maintained,” Caudill said.
Continue Reading
-

VVS Discord Stealer Using Pyarmor for Obfuscation and Detection Evasion
Executive Summary
This article details our technical analysis of VVS stealer, also styled VVS $tealer, including its distributors’ use of obfuscation and detection evasion.
The stealer is written in Python and targets Discord…
Continue Reading
-

St. Louis Symphony lures new audiences with hybrid concerts
Concertgoers interested in hearing the music of Tina Turner, Dolly Parton or Simon and Garfunkel have a new place to go — Powell Hall, home of the St. Louis Symphony Orchestra.
A spate of newly announced…
Continue Reading
-

Experts warn mental health chatbots can cause harm even as demand grows amid a shortage of therapists
But the laws, all passed this year, don’t fully address the fast-changing landscape of AI software development. And app developers, policymakers and mental health advocates say the resulting patchwork of state laws isn’t enough to…
Continue Reading