
An artificial intelligence replica of Val Kilmer, the Hollywood icon who died from pneumonia in 2025, will star in the forthcoming indie film As Deep as the Grave. Variety reported the…
Author: admin
-
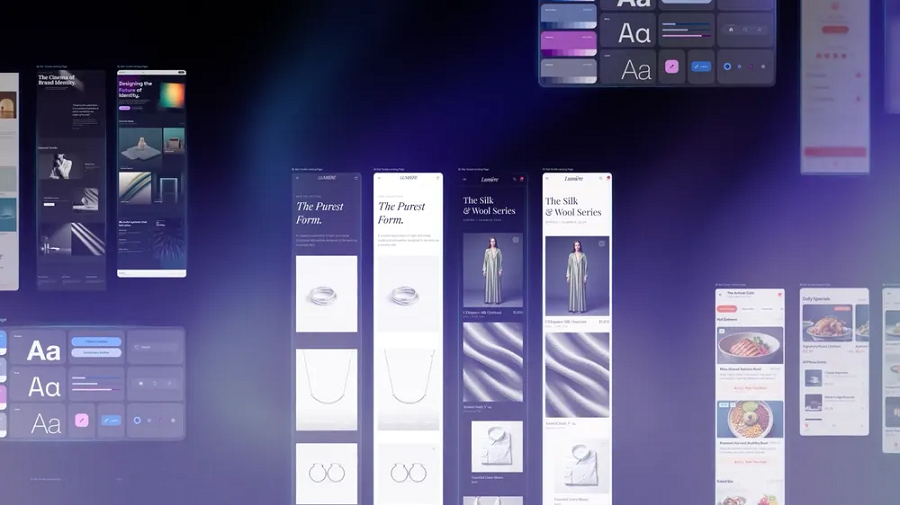
Google upgrades its Stitch AI interface development tool
Google LLC today released a new version of Stitch, an artificial intelligence tool that can generate user interfaces for websites and mobile apps.
Shares of graphic design software maker Figma Inc. declined more than 4% on the news. The…
Continue Reading
-

No Good Vibrations Here: The Beach Boys and John Stamos in Hot Water Over SeaWorld Gig
For Immediate Release:
March 19, 2026Contact:
David Perle 202-483-7382Orlando, Fla. –
Have mercy! Bearing…
Continue Reading
-
Conference League quarter-final ties confirmed: Dates and kick-off times – UEFA.com
- Conference League quarter-final ties confirmed: Dates and kick-off times UEFA.com
- Glasner reveals Crystal Palace plan to ‘make most’ of three-week break London Evening Standard
- From London to Athens: Interesting confrontation in the quarterfinals…
Continue Reading
-

Type 1 Diabetes Linked to Significantly Higher Dementia Risk
While diabetes has long been linked to dementia, new research suggests that people with type 1 may face a significantly higher risk of eventual cognitive decline.According to a study published in Neurology, people with type 1 diabetes were…Continue Reading
-

Game Preview: Warriors at Detroit Pistons – 3/20/26 – NBA
- Game Preview: Warriors at Detroit Pistons – 3/20/26 NBA
- Golden State Warriors vs Detroit Pistons Odds, Spread, and Prediction TheLines.com
- Golden State Warriors vs Detroit Pistons Prediction, 3/20/2026 Preview and Pick Doc’s Sports
- Pistons vs….
Continue Reading
-

AC/DC guitarist Stevie Young hospitalised ahead of Buenos Aires concerts | AC/DC
AC/DC guitarist Stevie Young has been hospitalised in Buenos Aires after feeling unwell, just days before the band’s scheduled sold-out concerts in the city.
According to the event’s promoter, the guitarist for the legendary Australian rock…
Continue Reading
-

Nets’ Michael Porter Jr. to miss at least 2-3 weeks with a strained left hamstring
Michael Porter Jr. is averaging a career-high 24.2 points, 7.1 rebounds and 3.0 assists in his first season in Brooklyn.
NEW YORK (AP) — Michael Porter Jr. will miss at least two weeks with a strained left hamstring, perhaps leaving the…
Continue Reading
-

Famous night for Nottingham Forest in a season to otherwise forget
While the Europa League will continue to offer respite from an otherwise difficult season for Forest, the additional games present challenges.
Forest will take on Porto in the quarter-final on 9 and 16 April, welcome Aston Villa to the City Ground…
Continue Reading
