I am deeply saddened at the passing of Seán Ó Sé.
A legend, Seán was an iconic figure in Irish education, culture and traditional music who had an extraordinary impact on Irish music and song. He was also an outstanding school principal and…

I am deeply saddened at the passing of Seán Ó Sé.
A legend, Seán was an iconic figure in Irish education, culture and traditional music who had an extraordinary impact on Irish music and song. He was also an outstanding school principal and…

When Team USA takes the ice at the 2026 Winter Olympics in Milano-Cortina, the roster will look like a…

LOGAN – Utah State University Opera Theatre’s November production of “Natasha, Pierre & The Great Comet of 1812” sold out seven performances, earning praise from audiences and local media while showcasing the depth of student talent and…

View of business offices and under construction residential apartments in Karachi, Pakistan September 23, 2025 Photo: Reuters
A gradual increase in…

Take a scroll down memory lane with your personalized Nintendo Switch™ 2025 Year in Review!
Once you’ve checked out all your stats, you can choose your favorite game of the year. And don’t forget to share your gaming story with others and…
Inside the new Lakewood Medical Offices, patients and staff will find 65 exam rooms, compared to 58 in the old building, meeting the need for one of Kaiser Permanente’s busiest primary care locations in Colorado. Larger exam rooms will add comfort and room for new medical technologies.
The new building doubles the number of urgent care exam rooms to 36 — which can help patients get urgent care more quickly.
With a newly designed flow through building, patients will be able to navigate services more easily.
The new Lakewood Medical Offices will also have more services available under a single roof. Services available will include:
The offices have new staff spaces for collaboration, as well as a lounge, conference center, gym, and lactation rooms.
The new Lakewood Medical Offices will be one of the first-of-their-kind, built using a new environmentally-friendly construction method. Components were manufactured off-site, assembled into sections, and installed at the construction site. The method cut landfill waste by nearly 70% and reduced transportation emissions.
The facility is targeting LEED Gold certification.
Additional environmentally friendly features include:
The new Lakewood Medical Offices are part of a larger commitment from Kaiser Permanente, the state’s largest nonprofit health plan and a leading medical provider. The organization announced in July 2025 it was making its largest brick-and-mortar investment in Colorado in 15 years.
Kaiser Permanente opened replacement Parker and Pueblo North medical offices in 2025. The organization is also planning to rebuild its Westminster Medical Offices — featuring an urgent care center and Kaiser Permanente’s first outpatient surgery center in Denver’s North Metro area — by 2028.
Combined, these investments are designed to give Kaiser Permanente members in Colorado more choice, convenience, and access to high-quality care across the Front Range.

Mon 12/1/2026
Minister News
.H. Sheikh Abdullah bin Zayed Al Nahyan, Deputy Prime Minister and Minister of Foreign…

LOUDONVILLE, NY – Siena Athletics has named Riley Powell to the position of Saints Assistant Athletic…
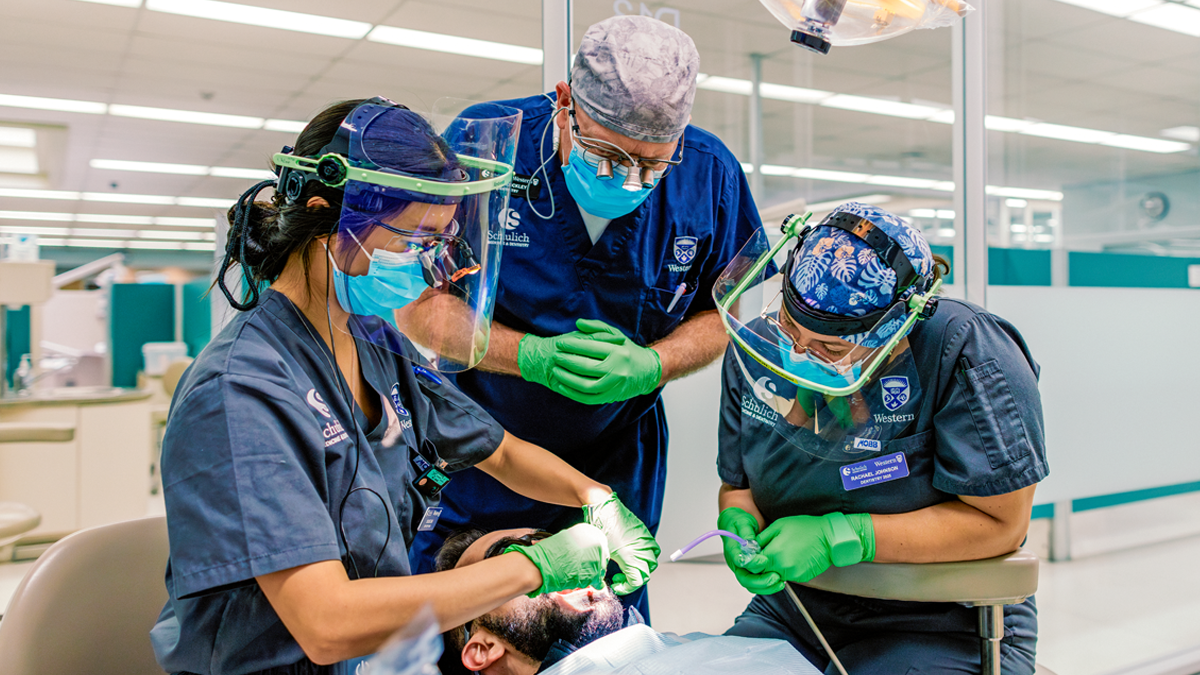
New federal investment will strengthen community partnerships, enhance clinic operations and prepare graduates for a changing dental care landscape