The projected fair value for Aalberts is €56.97 based on 2 Stage Free Cash Flow to Equity
Current share price of €28.64 suggests Aalberts is potentially 50% undervalued
The €37.75 analyst price target for AALB is 34% less than our estimate of fair value
How far off is Aalberts N.V. (AMS:AALB) from its intrinsic value? Using the most recent financial data, we’ll take a look at whether the stock is fairly priced by projecting its future cash flows and then discounting them to today’s value. We will take advantage of the Discounted Cash Flow (DCF) model for this purpose. Models like these may appear beyond the comprehension of a lay person, but they’re fairly easy to follow.
Remember though, that there are many ways to estimate a company’s value, and a DCF is just one method. Anyone interested in learning a bit more about intrinsic value should have a read of the Simply Wall St analysis model.
Trump has pledged to “unleash” American oil and gas and these 15 US stocks have developments that are poised to benefit.
We are going to use a two-stage DCF model, which, as the name states, takes into account two stages of growth. The first stage is generally a higher growth period which levels off heading towards the terminal value, captured in the second ‘steady growth’ period. To begin with, we have to get estimates of the next ten years of cash flows. Where possible we use analyst estimates, but when these aren’t available we extrapolate the previous free cash flow (FCF) from the last estimate or reported value. We assume companies with shrinking free cash flow will slow their rate of shrinkage, and that companies with growing free cash flow will see their growth rate slow, over this period. We do this to reflect that growth tends to slow more in the early years than it does in later years.
A DCF is all about the idea that a dollar in the future is less valuable than a dollar today, and so the sum of these future cash flows is then discounted to today’s value:
2026
2027
2028
2029
2030
2031
2032
2033
2034
2035
Levered FCF (€, Millions)
€268.5m
€298.1m
€319.8m
€337.9m
€353.0m
€366.0m
€377.4m
€387.7m
€397.2m
€406.1m
Growth Rate Estimate Source
Analyst x4
Analyst x4
Est @ 7.28%
Est @ 5.64%
Est @ 4.49%
Est @ 3.68%
Est @ 3.12%
Est @ 2.72%
Est @ 2.45%
Est @ 2.25%
Present Value (€, Millions) Discounted @ 7.2%
€250
€259
€259
€255
€249
€241
€231
€222
€212
€202
(“Est” = FCF growth rate estimated by Simply Wall St) Present Value of 10-year Cash Flow (PVCF) = €2.4b
After calculating the present value of future cash flows in the initial 10-year period, we need to calculate the Terminal Value, which accounts for all future cash flows beyond the first stage. The Gordon Growth formula is used to calculate Terminal Value at a future annual growth rate equal to the 5-year average of the 10-year government bond yield of 1.8%. We discount the terminal cash flows to today’s value at a cost of equity of 7.2%.
Present Value of Terminal Value (PVTV)= TV / (1 + r)10= €7.6b÷ ( 1 + 7.2%)10= €3.8b
The total value, or equity value, is then the sum of the present value of the future cash flows, which in this case is €6.2b. The last step is to then divide the equity value by the number of shares outstanding. Compared to the current share price of €28.6, the company appears quite undervalued at a 50% discount to where the stock price trades currently. The assumptions in any calculation have a big impact on the valuation, so it is better to view this as a rough estimate, not precise down to the last cent.
ENXTAM:AALB Discounted Cash Flow December 7th 2025
We would point out that the most important inputs to a discounted cash flow are the discount rate and of course the actual cash flows. You don’t have to agree with these inputs, I recommend redoing the calculations yourself and playing with them. The DCF also does not consider the possible cyclicality of an industry, or a company’s future capital requirements, so it does not give a full picture of a company’s potential performance. Given that we are looking at Aalberts as potential shareholders, the cost of equity is used as the discount rate, rather than the cost of capital (or weighted average cost of capital, WACC) which accounts for debt. In this calculation we’ve used 7.2%, which is based on a levered beta of 1.292. Beta is a measure of a stock’s volatility, compared to the market as a whole. We get our beta from the industry average beta of globally comparable companies, with an imposed limit between 0.8 and 2.0, which is a reasonable range for a stable business.
Check out our latest analysis for Aalberts
Strength
Weakness
Opportunity
Threat
Although the valuation of a company is important, it is only one of many factors that you need to assess for a company. It’s not possible to obtain a foolproof valuation with a DCF model. Rather it should be seen as a guide to “what assumptions need to be true for this stock to be under/overvalued?” If a company grows at a different rate, or if its cost of equity or risk free rate changes sharply, the output can look very different. Can we work out why the company is trading at a discount to intrinsic value? For Aalberts, we’ve compiled three further aspects you should explore:
Risks: We feel that you should assess the 3 warning signs for Aalberts we’ve flagged before making an investment in the company.
Management:Have insiders been ramping up their shares to take advantage of the market’s sentiment for AALB’s future outlook? Check out our management and board analysis with insights on CEO compensation and governance factors.
Other High Quality Alternatives: Do you like a good all-rounder? Explore our interactive list of high quality stocks to get an idea of what else is out there you may be missing!
PS. Simply Wall St updates its DCF calculation for every Dutch stock every day, so if you want to find the intrinsic value of any other stock just search here.
Have feedback on this article? Concerned about the content?Get in touch with us directly. Alternatively, email editorial-team (at) simplywallst.com.
This article by Simply Wall St is general in nature. We provide commentary based on historical data and analyst forecasts only using an unbiased methodology and our articles are not intended to be financial advice. It does not constitute a recommendation to buy or sell any stock, and does not take account of your objectives, or your financial situation. We aim to bring you long-term focused analysis driven by fundamental data. Note that our analysis may not factor in the latest price-sensitive company announcements or qualitative material. Simply Wall St has no position in any stocks mentioned.
Wondering if American Bitcoin at around $2.23 is a bargain or a value trap? You are not alone. This article is here to unpack what the market is really pricing in.
Despite all the hype around crypto infrastructure, the stock has been hammered recently, dropping about 47.4% over the last week, 51.9% over the last month, and 65.2% year to date. This has clearly reset expectations and risk appetite.
Those sharp moves have come alongside broader volatility in Bitcoin related names, shifting regulatory headlines around digital assets, and ongoing debates over the sustainability of crypto mining economics. At the same time, traders have been repricing high beta, speculative tech plays as interest rate expectations and liquidity conditions evolve.
Against that backdrop, American Bitcoin currently scores a 4 out of 6 on our valuation checks. This suggests it screens as undervalued on most, but not all, of our metrics. Next we will walk through those methods one by one before finishing with a more holistic way to think about what this stock is really worth.
American Bitcoin delivered 0.0% returns over the last year. See how this stacks up to the rest of the Software industry.
A Discounted Cash Flow model estimates what a business is worth by projecting the cash it can generate in the future and then discounting those cash flows back to today in $ terms.
For American Bitcoin, the latest twelve month Free Cash Flow is about $26.4 Million, and the 2 Stage Free Cash Flow to Equity model assumes this will grow rapidly over the next decade. Simply Wall St uses analyst estimates for the next few years, then extrapolates beyond that, with projected Free Cash Flow rising from roughly $45.7 Million in 2026 to about $231.6 Million by 2035 as growth gradually slows.
When these future cash flows are discounted back to today, the model arrives at an intrinsic value of roughly $2.99 per share. The current share price is about $2.23, so according to this DCF the stock is trading at a 25.3% discount. This indicates potential upside if the projected cash flow path occurs.
Result: UNDERVALUED
Our Discounted Cash Flow (DCF) analysis suggests American Bitcoin is undervalued by 25.3%. Track this in your watchlist or portfolio, or discover 907 more undervalued stocks based on cash flows.
ABTC Discounted Cash Flow as at Dec 2025
Head to the Valuation section of our Company Report for more details on how we arrive at this Fair Value for American Bitcoin.
For a company that is generating profits, the price to earnings, or PE, ratio is often the cleanest way to gauge what the market is willing to pay for each dollar of earnings. It naturally ties valuation to the bottom line, which tends to be more durable than short term swings in revenue or book value, especially in a cyclical, capital intensive space like Bitcoin mining.
What counts as a reasonable PE depends on how fast investors expect earnings to grow and how risky they perceive those earnings to be. Higher growth and more predictable cash flows usually justify a higher PE, while volatile or uncertain earnings deserve a discount. Right now, American Bitcoin trades on a PE of about 12.4x, which is far below the broader Software industry average of roughly 31.5x and well under a peer group that averages around 98.3x. This suggests the market is heavily discounting its earnings.
Simply Wall St goes a step further with its Fair Ratio, a proprietary view of what PE multiple a stock should trade on after accounting for its earnings growth outlook, risk profile, profit margins, industry characteristics and market capitalization. This is more informative than a simple peer or sector comparison, because it adjusts for whether American Bitcoin truly deserves a premium or discount. On that basis, the shares appear to be trading below their Fair Ratio. This points to a market price that does not fully reflect the company’s fundamentals.
Result: UNDERVALUED
NasdaqCM:ABTC PE Ratio as at Dec 2025
PE ratios tell one story, but what if the real opportunity lies elsewhere? Discover 1452 companies where insiders are betting big on explosive growth.
Earlier we mentioned that there is an even better way to understand valuation, so let us introduce you to Narratives, a simple way to connect the story you believe about a company with the numbers that sit behind its fair value. A Narrative is your own perspective on American Bitcoin, captured as assumptions about its future revenue, earnings and profit margins, which then flow into a financial forecast and finally into an estimated fair value per share. On Simply Wall St, millions of investors build and share Narratives on the Community page, making it easy and accessible to see how different stories translate into different valuations and buy or sell decisions by comparing each Narrative’s Fair Value to today’s Price. Narratives are also dynamic, so when new information like earnings releases or major news arrives, the numbers and fair value estimates can update to reflect the latest outlook. For example, one American Bitcoin Narrative on the platform might see aggressive growth and a high fair value while another assumes muted growth and a much lower fair value, giving you a clear sense of the range of reasonable outcomes.
Do you think there’s more to the story for American Bitcoin? Head over to our Community to see what others are saying!
NasdaqCM:ABTC Earnings & Revenue History as at Dec 2025
This article by Simply Wall St is general in nature. We provide commentary based on historical data and analyst forecasts only using an unbiased methodology and our articles are not intended to be financial advice. It does not constitute a recommendation to buy or sell any stock, and does not take account of your objectives, or your financial situation. We aim to bring you long-term focused analysis driven by fundamental data. Note that our analysis may not factor in the latest price-sensitive company announcements or qualitative material. Simply Wall St has no position in any stocks mentioned.
Companies discussed in this article include ABTC.
Have feedback on this article? Concerned about the content? Get in touch with us directly. Alternatively, email editorial-team@simplywallst.com
Before meeting for this interview, Tom Pickett opened Headspace, the app best known for mindfulness, and did a “breathing exercise” to “reset [his] brain”.
Usually he uses it at night. “My mind latches on to something before I go to bed, then I just can’t get it out of my head and I end up having poor sleep,” he says.
Since taking over as chief executive of Headspace in August last year, moving from US food delivery company, DoorDash, the 57-year-old, who has no formal experience in mental healthcare, has had a lot to get his head around. Over Zoom from his home office in San Francisco, Pickett has the look of a tech executive, with neat hair and a three-quarter zip fleece. In the background is a small sign: “Get Sh*t Done”.
“The healthcare industry is definitely complex. Every time you think you understand everything, you find that there’s a lot of nuances. It’s a constant learning process.”
Describing US-based Headspace as healthcare is a departure from its origins as a meditation app created by former Buddhist monk Andy Puddicombe and marketing executive Richard Pierson in 2010. It served up short digital programmes to busy professionals, with large employers, including Google, among the first to offer it to staff.
Four years ago, Headspace merged with Ginger, a mental health app focused more on coaching therapy and psychiatry, in a deal that valued the combined companies at $3bn. This broadened the product range to include access to virtual therapists, advice and workshops on topics such as mood management and insomnia. Headspace’s founders left in 2022, although Puddicombe’s voice continues to guide many of its meditations.
Since then, the company has intensified its push from consumers to employers and private health plans. More than 5.8mn people have access to Headspace through staff benefits provided by more than 4,500 organisations worldwide. In the new year, a deal with health insurer Cigna, will make the app accessible to an additional 7mn members in the US, through 18,000 employers’ health plans.
Overstretched public services and worsening mental health have created a gap for businesses to step in. But cynics see the type of digital service offered by Headspace as a cheap way to burnish the wellness credentials of organisations with long-hour cultures.
“This is one of the biggest challenges of our time,” says Pickett. “You can’t listen to the news without somebody talking about mental health, and increasingly so for the younger generations.”
The old model of “sending people to a therapist, a one-on-one human interaction” seemed inefficient to Pickett, who previously worked at Google, primarily for YouTube, for a decade.
“It was quite surprising to me . . . that there really isn’t a technology play in this space . . . We need to embrace technology. And if the primary modality of mental health is talking, then conversational AI has to have a big [part] in terms of how we solve this.”
Getting this right is among Pickett’s most important responsibilities. There have already been allegations of a chatbot encouraging one teenager to take his own life, and others urging users to self-harm. Mustafa Suleyma, Microsoft’s head of AI, has warned of “AI psychosis”, describing those who believe the technology is a God or lover.
“People are using AI tools that weren’t built for mental health,” warns Pickett. “General-use chatbots [are] built to do a lot of things. And they’re amazing. But they were not designed to take somebody who maybe has an acute mental illness and support them through a difficult time.”
Last year Pickett scaled back Headspace’s full-time therapists, moving them to part-time and contractor roles, to cut costs. At about the same time he launched the chatbot Ebb, which on Monday is upgrading from text-only to voice. Pickett insists this service is not designed for serious mental health issues and the company still offers remote therapists. Chatbots help “everyday emotional regulation” with bouts of anxiety or sleeplessness. “That’s frankly what a large part of the population really needs . . . Something to talk to, to reflect, to help process their emotions.”
Pickett says the company has developed a safety system to identify high-risk language and escalate serious concerns for human clinician review. All messages are monitored for potential risks, including suicidal and homicidal ideation, self-harm, domestic violence, substance use, eating disorders and abuse of vulnerable populations. When the conversation takes a turn into this territory, Ebb directs the user to crisis care, and ends the conversation.
Research finds well-designed chatbots can help with mental health issues, and some users find it easier to open up to them. “There’s some really interesting things that are evolving around people’s openness to put things on the table with a conversational AI in a way that they might not have done with a human therapist,” says Pickett.
He is “bullish” on AI’s potential. But “we have to make sure we protect against the downside”. At stake is the company’s reputation. “We’ve got 15 years of a brand that we’ve built up, building user trust, and we really do not want to lose that.”
The wellbeing sector is fiercely competitive. Headspace has lower downloads and monthly average users than its larger rival, Calm, according to Sensor Tower, the market intelligence company. But new downloads for both have fallen. In the third quarter of this year, Sensor Tower says Headspace’s monthly average users were 12 per cent lower than a year earlier.
Headspace says the fall reflects a shift from direct subscriptions to employer and health plans. Pickett says that, despite economic uncertainty, employers are not pulling back from wellbeing benefits. “Mental health continues to be a top two or three issue for companies . . . I think most of them want a solution that’s more than . . . the ‘call up a phone number’ kind of model.”
He hopes more insurers will follow Cigna by adding Headspace to employer schemes. “Historically, the only thing health plans covered was clinical . . . With employers, you go 10,000, 50,000 100,000 at a time, but with health plans, you go millions at a time. Ultimately, that could become the biggest part of our business.”
As a private company, Headspace does not disclose detailed financial information. Pickett says last year it made “north of $200mn in revenue” and operates “ebitda profitable”.
Pickett gained an insight into mental health struggles at the start of his career, when he spent nine years in the navy as an F-18 pilot, including two deployments to the Gulf. Behind him is a model and a large photo of F-18 aircraft. “We were put in stressful situations — a bunch of 18-year-olds to largely 30-somethings, away from their families. Stress, anxiety, [and] later forms of depression were out there, and people were trying to figure out how to deal with that.” Then, the attitude was “suck it up”.
His time in the navy provided a “great learning opportunity for management”, he says.
That might have helped when he took over at Headspace, and oversaw job cuts. “There were still elements of the merger that we were cleaning up. Systems integration, two products that you’re pushing into one. There [were] some cultural differences.”
Employees, he realised, were drawn by “the mission” of improving mental health; it “really matters deeply to people who work there”.
That has led to some criticism on Glassdoor, the anonymous employer review site, that Headspace’s culture is more focused on numbers than on wellbeing. One former employee wrote: “It’s quite ironic that so much of their content centres around taking care of your mental health at work.”
Pickett says the company is now in “a much better place”. “I’m happy with the size . . . today [about 400 staff]. People are definitely pushed. We have a lot to do.”
An IPO might be a long-term goal but for now his focus is on building a “sustainable, healthy business”. “You want the flexibility to move and evolve and invest.”
A day in the life of Tom Pickett
6:45am Wake up and check my Oura smart ring stats — seven hours of actual sleep is the goal. I then grab a coffee for the road and head to our San Francisco office.
Morning This is my peak problem-solving and creative-thinking window, so I frontload the day with meetings and anything that requires sharp thinking and clear decision-making.
Lunch When I’m not travelling, I eat at the office and try to catch up with others, all while staying on top of emails and Slack messages.
Afternoon Some days are dedicated to strategy deep dives, others are all about product. I do “skip-level” meetings to keep a pulse on what is going on through the organisation, do customer calls and meet my direct reports.
Evening It’s very important to me to get home for family dinner. With four kids, three still at home, this is the time to connect as a family. Everyone’s at the table, sharing their day.
I’ll try to squeeze in a run or walk. The rest of the night is spent helping with homework while prepping for the next day’s meetings. We have a no TV rule during the week.
Before bed, I wind down with Headspace, shut down all screens, and give myself the best shot at hitting my seven-hour sleep target.
Simply sign up to the Chinese trade myFT Digest — delivered directly to your inbox.
Chinese exports to south-east Asia are growing at almost twice the rate of the past four years, as Donald Trump’s trade war pushes Beijing to tighten trade links with its neighbours.
Chinese exports to the six largest economies in south-east Asia — Indonesia, Singapore, Thailand, the Philippines, Vietnam and Malaysia — rose 23.5 per cent from $330bn to $407bn in the first nine months of the year compared with the same period last year, according to official import data from those countries collated for the Financial Times by ISI Markets.
Chinese exports to those countries have doubled over the past five years, while China’s trade surplus with the region hit an all-time high this year. The 2025 increase is nearly twice as high as the 13 per cent compound annual growth rate in the previous four years.
China has long been criticised for “dumping” cheap goods in markets such as south-east Asia, threatening local producers with unfairly low prices but “the general China shock that has been going on for a few years has been amplified through US tariff deflection this year”, said Roland Rajah, lead economist at the Lowy Institute think-tank.
Economists say the latest wave of exports could be tied to attempts to circumvent US tariffs on Chinese-made products, which have been hit by levies of around 47 per cent. This compares with levies of about 19 per cent across many countries in south-east Asia.
Some content could not load. Check your internet connection or browser settings.
The US has warned against companies trying to mask the origin of Chinese-made products by rerouting them through other countries to avoid higher tariffs, saying such goods could be hit by “transshipment” levies of as much as 40 per cent. It is unclear how this has worked in practice.
In an upcoming paper, Rajah calculates Chinese exports to south-east Asia rose by as much as 30 per cent in September compared with a year earlier, noting that the most recent wave is different from earlier surges.
“While they are crowding out other exporters to the region, much of what they are exporting is actually pro-growth,” he said, adding that his research suggests as much as 60 per cent of Chinese exports this year were components for products manufactured in the region that were exported to other markets.
For consumer goods, China has increasingly become the dominant supplier to south-east Asia, taking market share from other countries.
“China’s supply glut, especially in cheap consumer goods, demands new outlets, and south-east Asia is the most natural spillover market given its proximity, logistics and scale,” said Doris Liew, an economist who formerly worked at Malaysia’s Institute for Democracy and Economic Affairs.
One area this has been most evident is in autos, with south-east Asian drivers switching in droves from Japanese models including the likes of Toyota, Honda and Nissan, to affordable electric cars made by China’s BYD.
The market share of Japan’s producers fell to 62 per cent of car sales in south-east Asia’s six biggest markets in the first half of 2025, down from an average of 77 per cent in the 2010s, according to PwC. China has increased its share from negligible volumes to more than 5 per cent of 3.3mn annual car sales in those markets.
In attempts to protect domestic manufacturers from being undercut by cheaper Chinese imports, some south-east Asian countries have tightened import rules and considered tariffs on certain goods.
But Liew said such actions were “piecemeal” and “stop-gap measures”. “The fundamental lesson is unavoidable: south-east Asian manufacturers must upgrade or be squeezed out,” she said. “China’s industrial ecosystem is far more innovative.”
Roula Khalaf, Editor of the FT, selects her favourite stories in this weekly newsletter.
Ben & Jerry’s co-founder Ben Cohen has clashed with the boss of Unilever’s ice cream spin-off after being told to “hand over to a new generation”, escalating a feud over the direction of the activist brand.
The row threatens to overshadow the demerger of the Magnum Ice Cream Company from its FTSE 100 parent as its shares start trading in Amsterdam on Monday.
Peter ter Kulve, Magnum’s chief executive, said that Cohen and Jerry Greenfield were in their seventies and “at a certain moment they need to hand over to a new generation”.
Their “commitment to the brand, to the causes, has been immense, but at a certain moment you need to hand it over . . . we need to move on”, he told the Financial Times in comments that also pertained to trustees of Ben & Jerry’s charitable arm, Jeff Furman and Liz Bankowski.
Cohen and Greenfield have become increasingly vocal about their dissatisfaction with the direction of the brand they launched almost 50 years ago and sold to Unilever for $326mn in 2000.
That deal put in place an independent board to protect the brand’s social mission and integrity. It also allowed Unilever to choose the chief executive, but only a minority of directors.
The co-founders have accused Unilever, and now Magnum — the ice cream business being spun off by the consumer goods giant — of impeding its activism. Greenfield quit Unilever in protest in September.
Cohen said: “Unlike Magnum, I don’t think there is an age limit on campaigning for social justice and peace. This is another attempt to silence the social mission that we are all too familiar with, as Unilever attempts to wash their hands of Ben & Jerry’s through this IPO. But Ben & Jerry’s social mission has always been inseparable from the brand itself, and it is legally protected.”
Greenfield, Furman and Bankowski did not respond to requests for comment.
Shares in the maker of Magnum, Carte D’Or, Cornetto and Solero will start trading on Monday in Amsterdam following the demerger from Unilever. Secondary listings in New York and London will take place later in the week.
Magnum will be the world’s largest ice cream company, with more than a fifth of the global market and annual revenues of €8bn. It is expected to perform better as a standalone company than as part of Unilever, which will retain a 19.9 per cent stake.
Magnum will inherit the campaigning Cohen, its most vocal employee, who has called on the parent to “free” the Chunky Monkey and Cookie Dough maker, and tried to raise funds to buy back Ben & Jerry’s.
Magnum also inherits a long-running legal spat with the Ben & Jerry’s board over its powers to define the company’s direction.
The board has accused Unilever of blocking its call for a ceasefire in Gaza, preventing it from supporting Palestinian refugees, and ousting David Stever as chief executive of Ben & Jerry’s earlier this year for failing to comply with Unilever’s attempts to silence the brand.
As the disputes drag on, Ter Kulve is cracking down on governance at the board and the brand’s charitable arm, the Ben & Jerry’s Foundation, which was funded by Unilever, and now Magnum.
Magnum said last month following an external investigation that Anuradha Mittal, chair of the brand’s independent board, “no longer meets the criteria” to serve but did not say why. People familiar with the matter said the investigation had uncovered conflicts of interest, again without disclosing what they were.
Counsel for the Ben & Jerry’s board said that Unilever’s “phantom allegations” were part of a campaign against Mittal due to her “efforts to protect the independence of Ben & Jerry’s under the merger agreement”.
The audit of the charity, meanwhile, uncovered “material deficiencies” in financial controls, governance and compliance, including conflicts of interest.
Ter Kulve said: “A significant amount of money, €5mn to €6mn a year goes to the foundation. I can’t continue to fund [the foundation] unless we basically have complied with the conclusions of the audit, and we’re working on that.”
Underlining his ambition for Ben & Jerry’s to break from the past, ter Kulve said he planned to expand the brand beyond its traditional tubs into sticks and ice cream sandwiches next year to capitalise on growing health awareness and consumer preference for smaller portion sizes.
“That was the big opportunity when you take a brand like Ben and Jerry’s . . . they were stuck in the pint [tub] for a very long time.”
Unilever announced it would spin off its ice cream division last year as part of a plan to slim down the sprawling multinational through lay-offs and divestments. Ter Kulve has set a medium-term organic sales growth target of 3 per cent, which is considered ambitious by analysts.
Barclays forecast a potential share price range of €20.78 to €21.46, based on an estimated equity value of €10.2bn to €10.5bn.
SAN FRANCISCO and SUZHOU, China, Dec. 6, 2025 /PRNewswire/ — Innovent Biologics, Inc. (“Innovent”) (HKEX: 01801), a world-class biopharmaceutical company that develops, manufactures and commercializes high quality medicines for the treatment of oncology, cardiovascular and metabolic, autoimmune, ophthalmology and other major diseases, announces that seven of its innovative products have been included in the updated 2025 National Reimbursement Drug List (NRDL). This list features a new indication of TYVYT® (sintilimab injection), and first-time inclusions of SYCUME® (teprotumumab N01 injection, a recombinant anti-IGF-1R antibody), Limertinib (EGFR TKI), Dupert® (fulzerasib, KRAS G12C inhibitor), DOVBLERON® (taletrectinib, ROS1 inhibitor), Retsevmo® (selpercatinib, RET inhibitor), and Jaypirca® (pirtobrutinib, BTK inhibitor). The updated NRDL will be officially effective from January 1, 2026.
Dr. Michael Yu, Founder, Chairman of the Board and CEO of Innovent, stated: “We are pleased with the NRDL inclusion of our seven innovative therapies this year. These therapies cover key disease areas that pose substantial public health challenges in China—particularly oncology (including lung, liver, gastric, esophageal, gynecological cancers, and hematological malignancies) as well as cardiovascular and metabolic (CVM) diseases. Their inclusion will help broaden patients’ access to and enhance their affordability of these medications, ultimately benefiting more individuals and families across the country. As a company with the mission of ’empowering patients worldwide with affordable, high-quality biopharmaceuticals’, Innovent continues to invest in pioneering treatments across oncology, CVM, autoimmune and ophthalmology—areas of significant societal need. We remain committed to our patient-centered approach, leveraging our innovation and product capabilities to further improve drug affordability and accessibility, so that high-quality medicines can reach and benefit more patients and their families as soon as possible. We are proud to contribute to better care for our patients.”
TYVYT® (sintilimab injection)
TYVYT® (sintilimab injection) is a PD-1 immunoglobulin G4 monoclonal antibody co-developed by Innovent and Eli Lilly and Company. In China, sintilimab has been approved for eight indications and two more NDAs are under review by the NMPA, including squamous non-small cell lung cancer (NSCLC), non-squamous NSCLC, liver cancer, gastric cancer, esophageal cancer, endometrial cancer and Hodgkin’s lymphoma[i].
In the updated NRDL, the eighth indication of TYVYT®(sintilimab injection) is newly included, in combination with fruquintinib for the treatment of patients with advanced endometrial cancer with Mismatch Repair proficient (pMMR) tumors that have failed prior systemic therapy and are not candidates for curative surgery or radiation. This new indication addresses a critical gap in treatments available for advanced endometrial cancer patients with limited responses to traditional therapies.
SYCUME® (teprotumumab N01 injection)
SYCUME® (teprotumumab N01 injection) is a recombinant anti-insulin-like growth factor 1 receptor (IGF-1R) antibody developed by Innovent. SYCUME® blocks the activation of IGF-1R signaling pathway, consequently improving clinical manifestations such as proptosis, inflammation and diplopia, thus enhancing quality of life in patients with thyroid eye disease (TED)[ii].
In the updated NRDL, SYCUME®(teprotumumab N01 injection) is newly listed for moderate-to-severe thyroid eye disease. SYCUME®(teprotumumab N01 injection) is China’s first approved IGF-1R antibody drug, and this groundbreaking non-invasive therapy redefines the standard of care and serves the unmet needs for thyroid eye disease over past 70 years in China. The NRDL inclusion will bring this world-class novel treatment option to Chinese patients with thyroid eye disease and significantly enhance patient accessibility and affordability.
Limertinib
Limertinib is a third-generation EGFR TKI in collaboration with ASK Pharm, and Innovent holds exclusive commercialization rights in Mainland China.[iii]
In the updated NRDL, limertinib is newly listed for: 1) the treatment of adult patients with locally advanced or metastatic EGFR T790M-mutated non-small cell lung cancer (NSCLC), who have previously experienced disease progression during or after treatment with EGFR TKI; and 2) the first-line treatment of adult patients with locally advanced or metastatic NSCLC carrying EGFR exon 19 deletions or exon 21 L858R mutations. Limertinib incorporates a unique naphthylamine group structure, which endows it with enhanced lipophilicity. This property ensures effective drug penetrate across the blood-brain barrier (BBB), thereby significantly reducing the risk of disease progression—specifically, the risk of disease progression in patients with brain metastases and the risk of intracranial disease progression. The NRDL inclusion of limertinib will provide a more effective treatment option for NSCLC patients with EGFR mutations.
Dupert® (fulzerasib)
Dupert® (fulzerasib) is a novel KRAS G12C inhibitor in collaboration with GenFleet Therapeutics, and Innovent holds exclusive development and commercialization rights in Greater China.[iv]
In the updated NRDL, Dupert®(fulzerasib) is newly listed for the treatment of advanced NSCLC adult patients harboring KRAS G12C mutation who have received at least one systemic therapy. The NRDL inclusion of Dupert®(fulzerasib) will provide a novel targeted therapy benefiting NSCLC patients harboring KRAS G12C mutation.
DOVBLERON® (taletrectinib)
DOVBLERON® (taletrectinib) is a novel next-generation ROS1 TKI in collaboration with Nuvation Bio China, a Nuvation Bio (NYSE: NUVB) Company, and Innovent holds exclusive commercialization rights in Greater China.[v]
In the updated NRDL, DOVBLERON® (taletrectinib) is newly listed for the treatment of adult patients with locally advanced or metastatic ROS1-positive NSCLC. The NRDL inclusion of DOVBLERON® (taletrectinib) will provide a potentially best-in-class therapy benefiting patients with locally advanced ROS1-positive NSCLC.
Retsevmo® (selpercatinib)
Retsevmo® (selpercatinib) is a selective and potent rearranged during transfection (RET) kinase inhibitor developed by Eli Lilly and Company and solely commercialized in Mainland China by Innovent.[vi]
In the updated NRDL, Retsevmo® (selpercatinib) is newly listed for the treatment of: 1) adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with a RET gene fusion, 2) adult and pediatric patients 12 years of age and older with advanced or metastatic medullary thyroid cancer (MTC) with a RET mutation who require systemic therapy, and 3) adult and pediatric patients 12 years of age and older with advanced or metastatic thyroid cancer with a RET gene fusion who require systemic therapy and who are radioactive iodine-refractory. Retsevmo® (selpercatinib) is the first RET inhibitor approved globally and its NRDL inclusion will bring an innovative therapy for NSCLC and thyroid cancer patients with a RET alteration.
Jaypirca® (pirtobrutinib)
Jaypirca® (pirtobrutinib) is a non-covalent (reversible) BTK inhibitor developed by Eli Lilly and Company and solely commercialized in Mainland China by Innovent. Jaypirca® (pirtobrutinib) is the first and only non-covalent (reversible) BTK inhibitor approved in the world.[vii]
In the updated NRDL, Jaypirca® is newly listed for the treatment of adult patients with relapsed or refractory mantle cell lymphoma after at least two types of systemic therapy, including a BTK inhibitor. Its NRDL inclusion will benefit heavily-treated MCL patients that previously received the treatment of a BTK inhibitor, addressing their unmet needs and further enhancing Jaypirca’s affordability for those patients.
About Innovent
Innovent is a leading biopharmaceutical company founded in 2011 with the mission to empower patients worldwide with affordable, high-quality biopharmaceuticals. The company discovers, develops, manufactures and commercializes innovative medicines that target some of the most intractable diseases. Its pioneering therapies treat cancer, cardiovascular and metabolic, autoimmune and eye diseases. Innovent has launched 17 products in the market. It has 1 new drug applications under regulatory review, 4 assets in Phase 3 or pivotal clinical trials and 15 more molecules in early clinical stage. Innovent partners with over 30 global healthcare companies, including Eli Lilly, Roche, Takeda, Sanofi, Incyte, LG Chem and MD Anderson Cancer Center.
Guided by the motto, “Start with Integrity, Succeed through Action” Innovent maintains the highest standard of industry practices and works collaboratively to advance the biopharmaceutical industry so that first-rate pharmaceutical drugs can become widely accessible. For more information, visit www.innoventbio.com, or follow Innovent on Facebook and LinkedIn.
Statements:
Innovent does not recommend the use of any unapproved drug (s)/indication (s).
Ramucirumab(Cyramza), Selpercatinib (Retsevmo) and Pirtobrutinib (Jaypirca) were developed by Eli Lilly and Company.
Forward-Looking Statements
This news release may contain certain forward-looking statements that are, by their nature, subject to significant risks and uncertainties. The words “anticipate”, “believe”, “estimate”, “expect”, “intend” and similar expressions, as they relate to Innovent, are intended to identify certain of such forward-looking statements. The Company does not intend to update these forward-looking statements regularly.
These forward-looking statements are based on the existing beliefs, assumptions, expectations, estimates, projections and understandings of the management of the Company with respect to future events at the time these statements are made. These statements are not a guarantee of future developments and are subject to risks, uncertainties and other factors, some of which are beyond the Company’s control and are difficult to predict. Consequently, actual results may differ materially from information contained in the forward-looking statements as a result of future changes or developments in our business, the Company’s competitive environment and political, economic, legal and social conditions.
The Company, the Directors and the employees of the Company assume (a) no obligation to correct or update the forward-looking statements contained in this site; and (b) no liability in the event that any of the forward-looking statements does not materialise or turn out to be incorrect.
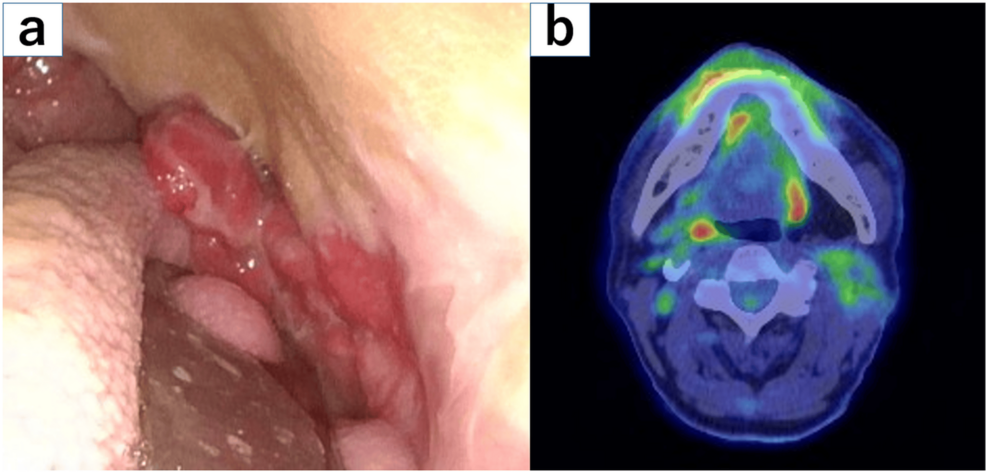
Treatment with fixed-duration epcoritamab-bysp (Epkinly) plus dose-attenuated rituximab (Rituxan) plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-mini-CHOP) appeared to be well tolerated and elicited responses in elderly patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL), according to data from arm 8 of the phase 1/2 EPCORE NHL-2 trial (NCT04663347).1
The data, which were presented at the 2025 ASH Annual Meeting, showed that at a median follow-up of 33.4 months (range, 2.8-41.1), the overall response rate (ORR) in all patients (n = 28) was 93%, which was comprised of a complete response (CR) rate of 86% and a partial response (PR) rate of 7%. The median time to response was 1.4 months (range, 1.1-2.7) and the median time to CR was 1.6 months (range, 1.2-8.1).
At 2 years, 79% (95% CI, 57%-91%) of patients remained in response to treatment with the regimen, and 79% (95% CI, 57%-91%) remained in CR. Of the 22 patients who completed therapy, 20 experienced a CR at the end of treatment (91%). At a median follow-up of 22.6 months after the end of treatment, 90% of the 20 patients remained in CR. Moreover, the 2-year progression-free (PFS) and overall survival (OS) rates were 76% (95% CI, 54%-89%) and 82% (95% CI, 62%-92%), respectively. The median PFS and OS were not reached, irrespective of International Prognostic Index score.
Examining Top Takeaway from the EPCORE NHL-2 Study of Epcoritamab in DLBCL
Fixed-duration epcoritamab plus R-mini-CHOP induced deep, rapid, and durable responses in elderly, frail patients with newly diagnosed DLBCL, with an ORR of 93% and a CR rate of 86%.
High MRD negativity rates (95%) and strong 2-year PFS (76%) and OS (82%) rates suggest meaningful long-term benefit despite high-risk baseline features.
The regimen was generally tolerable, with mostly manageable CRS and infection-related toxicities, supporting its potential role for patients unable to receive full-dose R-CHOP.
Of the 21 patients evaluable for minimal residual disease (MRD), 20 (95%) were negative at the time of the data cutoff date of September 21, 2025. At the first assessment, which was done on day 1 of cycle 3, 80% (n = 16) of patients achieved MRD negativity. Of the 4 MRD-positive patients at this assessment, 3 converted to MRD negativity by the second assessment, which was done on day 1 of cycle 6; 2 patients experienced subsequent progressive disease. Notably, MRD negativity was observed across all patient subgroups, including bulky disease (89%) and those with an IPI score ranging from 3 to 5 (93%).
“We can conclude that in combination with dose-attenuated chemotherapy, [epcoritamab] may have a role in the treatment of patients with historically poor outcomes,” Chan Cheah, MD, of the Sir Charles Gairdner Hospital and the University of Western Australia, in Nedlands, Australia, said during a presentation of the data.
Why add epcoritamab to R-mini-CHOP?
Although R-mini-CHOP is the standard of care (SOC) for patients with newly diagnosed DLBCL who are not able to receive the full dose, outcomes with the regimen remain suboptimal, Cheah explained. “It’s clear that better options for these patients are required,” he added. Previously, the CD3xCD20 bispecific antibody epcoritamab was found to be efficacious when administered as a monotherapy or paired with SOC in those with newly diagnosed DLBCL.
For example, findings from the phase 2 EPCORE DLBCL-3 trial (NCT05660967 ) indicated that single-agent epcoritamab induced durable responses in patients with newly diagnosed large B-cell lymphoma and comorbidities.2 Other findings from EPCORE NHL-2 showed that the combination of epcoritamab and R-mini-CHOP elicited an ORR of 89% and a CR rate of 82% in elderly patients with newly diagnosed DLBCL who could not receive the full dose of R-CHOP.3 At the meeting, Cheah shared follow-up data from EPCORE NHL-2.1
What did EPCORE NHL-2 evaluate?
The open-label, phase 1b/2 trial enrolled patients with newly diagnosed DLBCL, which could have been DLBCL not otherwise specified, T-cell or histocyte-rich DLBCL, high-grade B-cell lymphoma, or grade 3B follicular lymphoma. Patients had an ECOG performance status ranging from 0 to 2 and could not be eligible to receive full-dose R-CHOP because they were 75 years of age or older or 65 years of age or older with a comorbidity.
Twenty-eight patients received a fixed duration of subcutaneous epcoritamab at a dose of 48 mg once weekly for cycles 1 and 2 and every 3 weeks for cycles 3 to 6 and intravenous R-mini-CHOP, which comprised 375 mg/m2 of rituximab, 400 mg/m2 of cyclophosphamide, 25 mg/m2 of doxorubicin, 1 mg/m2 of vincristine—all given every 3 weeks—and 100 mg/day of prednisone, given on days 1 to 5 of each cycle from cycles 1 to 6. Epcoritamab was then given at 48 mg every 4 weeks for cycles 7 to 8.
ORR by investigator assessment served as the primary end point of the trial. Secondary end points included CR rate, duration of response, duration of CR, PFS, OS, MRD negativity, and safety.
In terms of baseline characteristics, the median patient age was 81 years (range, 74-90). Patients were not eligible to receive a full dose of anthracycline because of age older than 75 years (96%), hypertension requiring treatment (54%), diabetes mellitus (11%), or history of myocardial infarction (4%). Moreover, 43% of patients had an IPI score of 4 to 5 at screening, 39% had a bulky tumor of 7 cm or larger, and the majority had elevated lactate dehydrogenase (64%).
What was the safety profile of epcoritamab in this population?
Cheah noted that most adverse effects (AEs) were mild to moderate in severity, and no new safety concerns associated with the addition of epcoritamab to R-mini-CHOP presented with longer follow-up. The most frequent grade 3 or higher treatment-emergent adverse effects (TEAEs) were neutropenia (43%), serious infections (32%), and anemia 14%). Most grade 3 or higher serious infections were reported within the first 6 cycles of treatment, when R-mini-CHOP was being coadministered.
The most common TEAE was cytokine release syndrome (CRS), which occurred in 61% of patients; this effect was grade 1 for 32% of patients and grade 2 for 29% of patients. Time to first onset of CRS occurred at a median of 16 days (range, 8-17). Patients were treated with either tocilizumab (Actemra; 29%) or corticosteroids (14%), leading to CRS resolution across all patients affected. The median time to resolution was 2 days (range, 1-7). CRS led to treatment discontinuation in 1 patient. Cheah noted that almost all (90%) CRS events occurred in cycle 1 of treatment.
Additional common TEAEs occurring in 20% or more patients were neutropenia, serious infections, anemia, constipation, fatigue, hypokalemia, and fall. No patients experienced immune effector cell–associated neurotoxicity syndrome (ICANS) or tumor lysis syndrome (TLS).
TEAEs led to discontinuation of epcoritamab in 3 patients (11%), including 1 fatal event, and discontinuation of R-mini-CHOP in 6 patients (21%).
What is the significance of the updated EPCORE NHL-2 data?
“Despite an older population of newly diagnosed diffuse large B-cell lymphoma, the outcomes observed in arm 8 of the EPCORE NHL-2 evaluating fixed-duration epcoritamab plus R-mini-CHOP are encouraging,” Cheah stated in a news release issued by Genmab.4 “These results, along with those from other arms of the trial, support the potential for combinations of epcoritamab with standard of care treatment across a range of disease settings and patient populations.”
References
Cheah C, Ďuraš J, Belada D, et al. Epcoritamab + R-mini-CHOP results in 2-year remissions and high MRD negativity rates in elderly patients with newly diagnosed DLBCL: Results from the EPCORE NHL-2 trial. Presented at: 2025 ASH Annual Meeting; December 6-9, 2025; Orlando, FL. Abstract 64.
Vitolo U, Duell J, Burgues JMB, et al. Fixed-duration epcoritamab monotherapy induces high response and MRD-negativity rates in elderly patients with newly diagnosed large B-cell lymphoma (LBCL) and comorbidities: Results from EPCORE DLBCL-3. Presented at: 2025 ASH Annual Meeting; December 6-9, 2025; Orlando, FL. Abstract 63.
Leslie LA, Cheah CY, Morschhauser F, et al. Fixed-duration epcoritamab + R-mini-CHOP in patients with previously untreated diffuse large B-cell lymphoma ineligible for full-dose R-CHOP: Updated results from arm 8 of the Epcore NHL-2 trial. Blood. 2024;144(suppl 1):3106. doi: 10.1182/blood-2024-199652
Genmab press release. Genmab announces data from multiple clinical trials showing treatment with fixed-duration epcoritamab led to remissions in first-line diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL). News release. Genmab. December 6, 2025. Accessed December 6, 2025. https://ir.genmab.com/news-releases/news-release-details/genmab-announces-data-multiple-clinical-trials-showing-treatment
Federal Reserve Chair Jerome Powell is set to preside over the US central bank’s last monetary policy meeting of 2025 amid a divided board (JUSTIN SULLIVAN)
While the US Federal Reserve’s final interest rate meeting this year could see an unusual amount of division, financial markets view a third straight interest rate cut as nearly certain.
When the Fed last met in October, Chair Jerome Powell asserted that another rate cut in December was “not a foregone conclusion,” pointing to “strongly differing views” within the central bank.
Minutes from the Fed’s most recent meeting showed many officials expect a further uptick in underlying goods inflation as President Donald Trump’s tariffs bite.
But recent comments from leading Fed officials also reflected support for cutting again because of a weakening labor market, even though inflation is still above the Fed’s two percent target.
Next week’s outcome in the “deeply divided” Fed was “too close to call,” UniCredit said, also acknowledging that favorable comments from New York Fed bank chief John Williams towards a cut were a notable “intervention.”
“As one of the most senior members of the (Fed committee), it seems unlikely Williams would have said this without Powell’s prior approval,” UniCredit said.
Policymakers generally hold rates at a higher level to tamp down price increases, but a rapidly deteriorating jobs market could nudge them to slash rates further to boost the economy.
“Usually, as you get closer to a policy meeting, it becomes quite apparent and transparent what the Federal Open Market Committee is going to do,” said Nationwide Chief Economist Kathy Bostjancic, referring to the Fed’s rate-setting committee.
“This time is very different,” she told AFP late last month.
Financial markets rallied following Williams’ statement on November 21 that rates could go lower in the “near term.”
Futures markets currently show more than 87 percent odds that the Fed will cut rates to between 3.50 percent and 3.75 percent, according to CME FedWatch.
– Dearth of data –
The Fed moved into rate cutting mode this fall, with rate cuts both in September and October.
But a government shutdown from October 1 through November 12 sapped the central bank of most of the key data points for assessing whether inflation or employment is now the bigger priority.
The latest available government data showed the jobless rate crept up from 4.3 percent to 4.4 percent in September, even as hiring beat expectations.
While delayed publications on September’s economic conditions have trickled out, the US government has canceled full releases of October jobs and consumer inflation figures because the shutdown hit data collection.
Instead, available figures will be published with November’s reports, but only after the Fed’s upcoming rate meeting.
The US personal consumption expenditures price index rose to 2.8 percent on an annual basis in September, from 2.7 percent in August, according to delayed data released on Friday.
The “Fed faces a bit of a paradoxical situation,” said EY-Parthenon Chief Economist Gregory Daco. “The Fed says these decisions will be data-dependent, but there isn’t a lot of data to go on.”
Daco expects a “weak majority” to favor another interest rate cut, but believes there could be multiple dissents.
– Looking beyond Powell –
Besides Wednesday’s decision, the Fed will also release projections for its 2026 economic and monetary policy outlook.
Next year will already mark a period of significant change with the conclusion of Powell’s tenure as chair in May.
Trump, who has relentlessly criticized Powell for not cutting rates more aggressively, signaled this week that his chief economic adviser Kevin Hassett could succeed Powell.
Hassett has appeared to be in lockstep with Trump on key economic questions facing the Fed. But if appointed, Hassett could also face pressure from financial markets to buck the White House on interest rates if inflation worsens.
“The institutional constraints often end up leading appointees towards some level of political independence,” said Daco, noting decisions require a board majority.
Whomever Trump picks will need to be confirmed in the US Senate.
While UniCredit predicted “political interference will have a modest impact on Fed policy,” deeper consequences cannot be ruled out.
“We have not assumed Trump will get de-facto control of the Fed,” UniCredit said, adding that such an outcome is “a non-negligible risk.”
Wondering whether STAAR Surgical at around $25 a share is a bargain or a value trap? Let us unpack what the market is really pricing in and what the fundamentals say.
The stock is roughly flat over the last year at about 0.3% while still up 5.3% year to date, but those modest gains sit on top of a painful multi year slide of around 58% over three years and nearly 68% over five.
That kind of long term drawdown usually reflects shifting expectations around growth and competitive pressure in its niche of implantable lenses, as investors reassess how fast premium vision correction can scale. At the same time, renewed interest in specialized medical devices and structural demand for refractive surgery keeps STAAR on many watchlists, even without splashy headline catalysts.
On our high level checks, STAAR currently scores just 2 out of 6 for undervaluation, which suggests pockets of value but not a screaming deal on traditional metrics alone. Next, we will walk through the main valuation approaches that produce that score and outline a more insightful way to think about what the stock might really be worth by the end of this article.
STAAR Surgical scores just 2/6 on our valuation checks. See what other red flags we found in the full valuation breakdown.
A Discounted Cash Flow model estimates what a business is worth today by projecting its future cash flows and then discounting them back to the present using a required rate of return.
For STAAR Surgical, the latest twelve month Free Cash Flow is negative at about $42.7 Million, which reflects current investment and profitability challenges. Analysts project an improvement, with Free Cash Flow expected to reach $66 Million by 2029, and Simply Wall St then extrapolates further growth out to 2035 based on those analyst inputs.
When all projected cash flows are discounted back to today using a 2 Stage Free Cash Flow to Equity model, the intrinsic value comes out at around $37.33 per share. With the stock trading near $25, the DCF suggests the shares are roughly 32.0% undervalued and that the market may be skeptical that STAAR will fully deliver on these cash flow improvements.
Result: UNDERVALUED
Our Discounted Cash Flow (DCF) analysis suggests STAAR Surgical is undervalued by 32.0%. Track this in your watchlist or portfolio, or discover 911 more undervalued stocks based on cash flows.
STAA Discounted Cash Flow as at Dec 2025
Head to the Valuation section of our Company Report for more details on how we arrive at this Fair Value for STAAR Surgical.
For companies where earnings are volatile or negative, the Price to Sales ratio is often a cleaner way to think about valuation. It focuses on what investors are paying for each dollar of revenue rather than profit that can swing with short term spending decisions.
A higher or lower “normal” multiple tends to reflect how quickly a business can grow those sales and how risky that growth looks. Faster, more predictable growth usually aligns with a higher Price to Sales ratio, while slower or shakier growth is often closer to or below the market and industry averages.
STAAR Surgical currently trades on a Price to Sales ratio of about 5.47x, which is meaningfully above both the Medical Equipment industry average of roughly 3.37x and the peer average of around 3.37x. Simply Wall St’s Fair Ratio for STAAR is 3.79x, a proprietary estimate of the multiple the stock could trade on given its growth outlook, profitability, industry, size, and risk profile.
Because the Fair Ratio blends all of these fundamentals, it is a more nuanced benchmark than a simple peer or industry comparison. With STAAR’s current 5.47x sitting well above the 3.79x Fair Ratio, the stock screens as overvalued on this measure.
Result: OVERVALUED
NasdaqGM:STAA PS Ratio as at Dec 2025
PS ratios tell one story, but what if the real opportunity lies elsewhere? Discover 1450 companies where insiders are betting big on explosive growth.
Earlier we mentioned that there is an even better way to understand valuation, so let us introduce you to Narratives, a simple way to connect your view of a company with the numbers behind it. A Narrative is the story you believe about a business, expressed through assumptions about its future revenue, earnings and margins, which then flow through to a forecast and an estimated fair value. On Simply Wall St, Narratives are an easy, accessible tool on the Community page, where millions of investors can turn their perspective on a company into a structured financial view. By comparing your Narrative Fair Value to the current share price, you can quickly decide how STAAR Surgical aligns with your own investment view as a potential buy, hold or sell, and because Narratives update dynamically as new news, earnings or guidance comes in, your view remains current rather than static. For example, on STAAR Surgical today, one investor might see upside to a fair value around $28 on the back of a China recovery and new products, while another could take a more cautious stance closer to $16 if they doubt the rebound and worry more about competition and execution risk.
Do you think there’s more to the story for STAAR Surgical? Head over to our Community to see what others are saying!
NasdaqGM:STAA Community Fair Values as at Dec 2025
This article by Simply Wall St is general in nature. We provide commentary based on historical data and analyst forecasts only using an unbiased methodology and our articles are not intended to be financial advice. It does not constitute a recommendation to buy or sell any stock, and does not take account of your objectives, or your financial situation. We aim to bring you long-term focused analysis driven by fundamental data. Note that our analysis may not factor in the latest price-sensitive company announcements or qualitative material. Simply Wall St has no position in any stocks mentioned.
Companies discussed in this article include STAA.
Have feedback on this article? Concerned about the content? Get in touch with us directly. Alternatively, email editorial-team@simplywallst.com