NASA‘s James Webb Space Telescope has revealed that an exceptionally rare star duo in the Milky Way has a third companion — and it’s a monster.
The star system, named Apep after the…

NASA‘s James Webb Space Telescope has revealed that an exceptionally rare star duo in the Milky Way has a third companion — and it’s a monster.
The star system, named Apep after the…
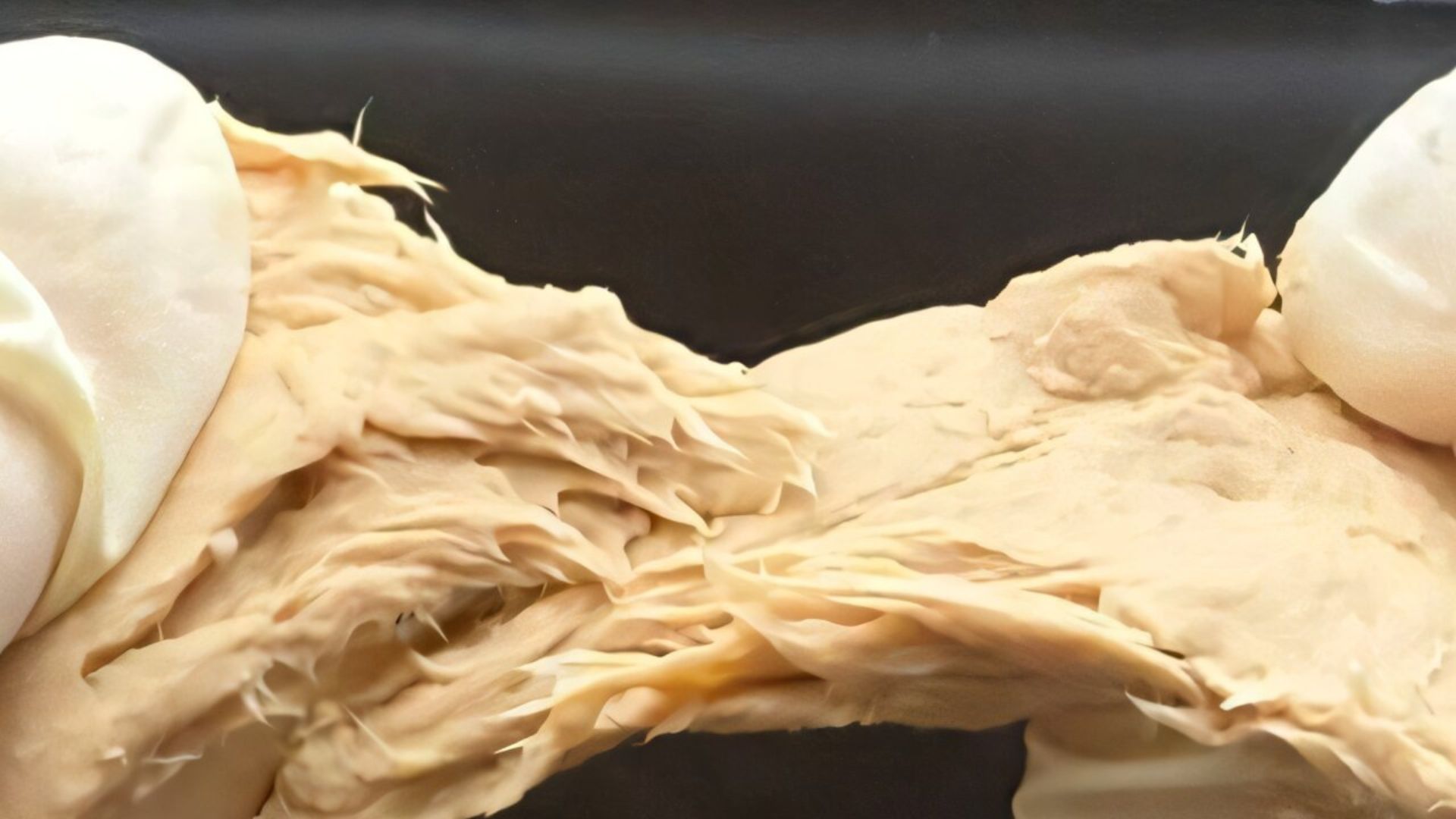
Forget lab-grown burgers. A new genetically tweaked fungus is here, and it’s expected to shape the future of sustainable food.
Researchers in China have unveiled a modified fungus that not only tastes like meat but also cuts production…

The European Space Agency’s Euclid mission-designed to map the geometry of the dark Universe with unprecedented precision-continues to deliver its first scientific insights. The Euclid Consortium has published a fresh set of seven…

Two of Jupiter’s Galilean moons cross the planet overnight, as their shadows race ahead. Which shadow will win?
By 9:30 P.M. EST, the shadows of Io…

EUMETSAT, Europe’s meteorological satellite agency, monitors the weather and climate from space. Based in Darmstadt, Germany, EUMETSAT provides its 30 member states with meteorological imagery and data that are essential for…


BEIJING — A research team has found a way to make certain rare earth materials, known for their bright and stable light, emit light when powered directly by electricity.
The study, co-conducted by researchers from…

A rare astronomical event will occur on Thursday, Nov. 20, 2025, as a new moon reaches its farthest distance from Earth until 2043. Occurring four hours before the moon reaches its new phase at 1:47 a.m. EST, according to Timeanddate.com,…
