Event Info
Here’s how to watch the 2025 TBC vs TBC broadcast on FloRugby. The 2025 TBC vs TBC broadcast starts on Oct 11, 2025. Stream or cast from your desktop, mobile or TV. Now available on Roku, Fire TV, Chromecast and Apple TV. Don’t forget…

Here’s how to watch the 2025 TBC vs TBC broadcast on FloRugby. The 2025 TBC vs TBC broadcast starts on Oct 11, 2025. Stream or cast from your desktop, mobile or TV. Now available on Roku, Fire TV, Chromecast and Apple TV. Don’t forget…
Nintendo has issued a statement refuting claims it has been lobbying the Japanese government about protecting its intellectual property against generative AI.
In a now-deleted message posted to his verified X account, Japanese…



Some exciting news emerged for chip stock Intel (INTC), particularly if the news proves accurate. But investors did not take it near so well as might be expected. Reports noted that Intel’s planned Panther Lake lineup may have leaked early,…
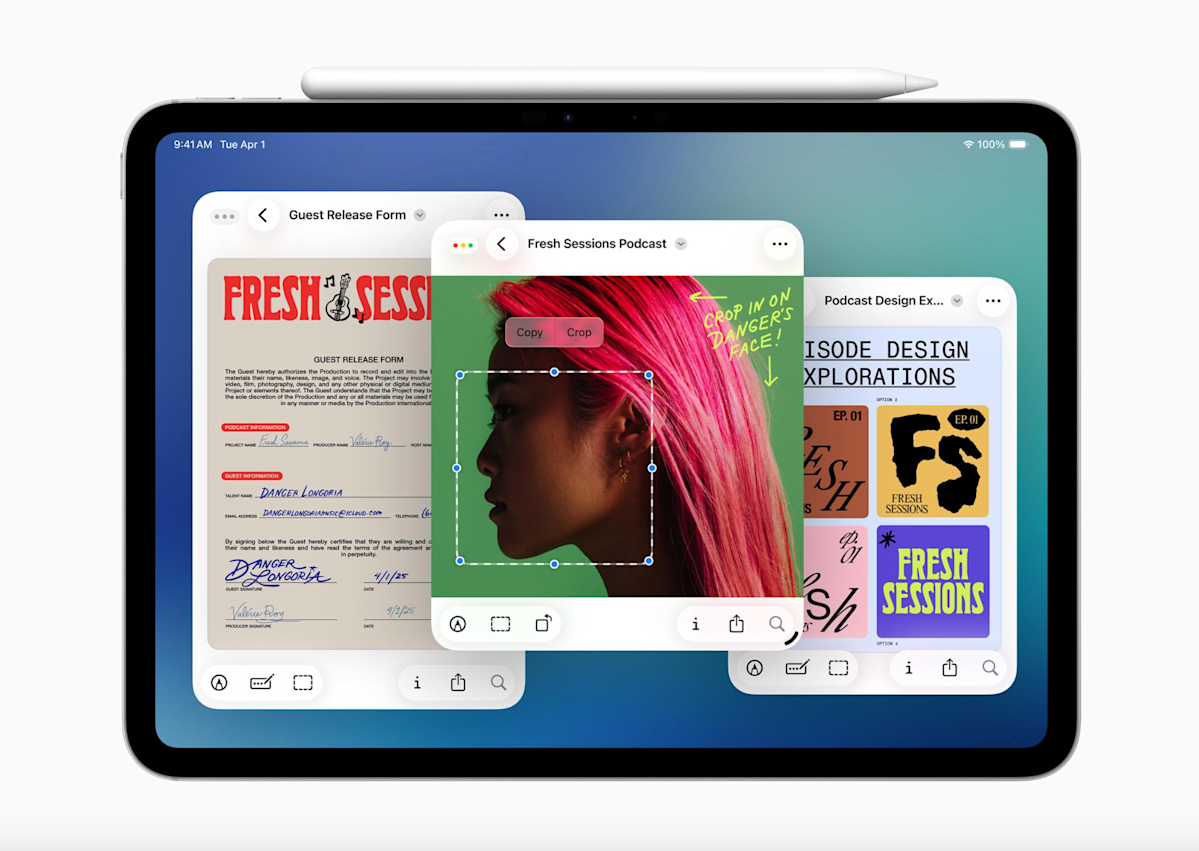
The reaction to iPadOS 26 , but some users have bemoaned the absence of the decade-old Slide Over multitasking feature. Well, Apple just announced that it’s coming back as part of iPadOS 26.1. It’s actually already available, though just as part…
Research led by the University of Michigan’s Kresge Hearing Research Institute and the University of Rochester illuminates the mechanisms through which humans can pick out and focus on single sounds in noisy…

Listen and Subscribe: Apple | Spotify
Sign up for our emails to never miss an interview, book excerpt, cover reveal, or book review.
On every First Taste Reading Series episode, a writer introduces their book and reads…
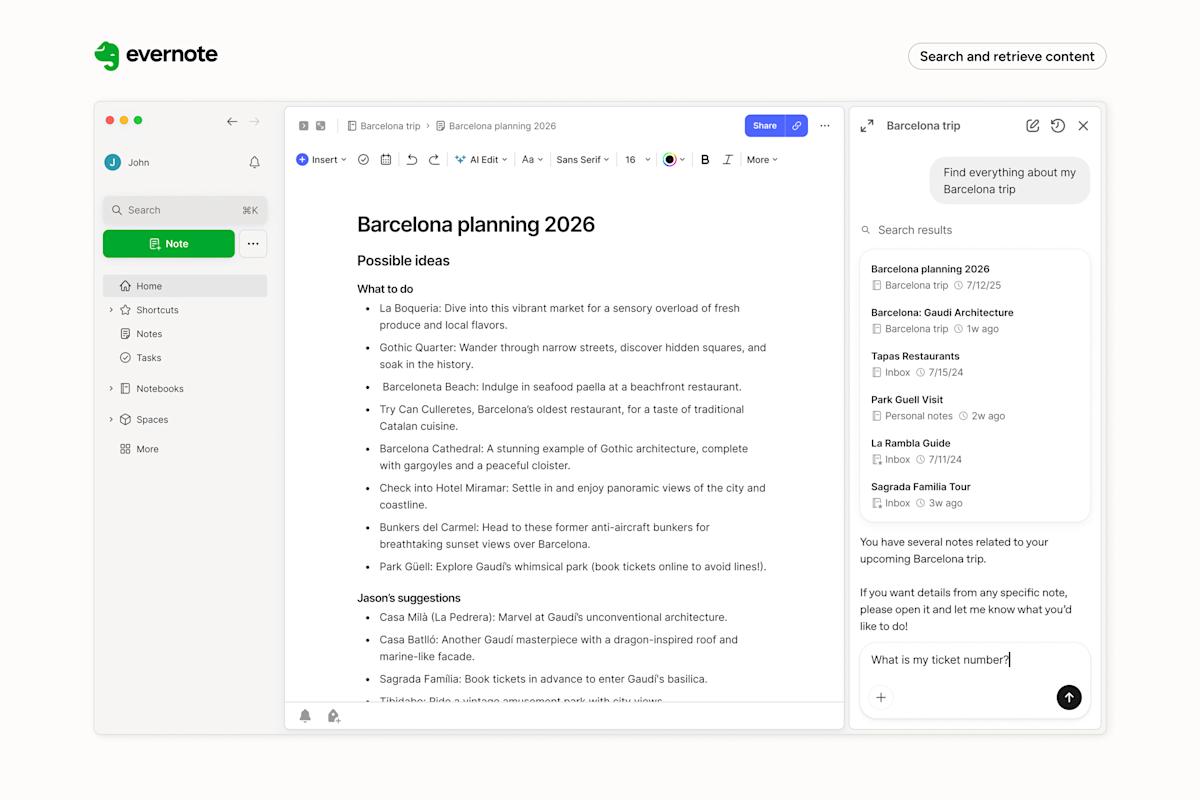
If you’re like me, it’s probably been a hot minute since you’ve thought about Evernote. For years, the note-taking app, once a darling of the App Store, faced declining popularity and profitability. The last time it grabbed headlines was in 2022…

For those who missed out on the past few years of ‘smart home’ gadgets, the Logitech POP buttons were introduced in 2018 as a way to control smart home devices using these buttons and a central hub. After a few years of…