
Capital One shares rose on Tuesday evening after the credit card issuer reported a sizeable quarterly beat, driven by improved credit quality performance. Revenue in the third quarter ended Sept. 30 increased 53% year over year to $15.36 billion, beating the LSEG-complied consensus estimate of $15.08 billion. Adjusted earnings per share increased 32% year over year to $5.95, exceeding the $4.37 estimate, LSEG data showed. This adjusted figure excludes a $1.12 after-tax diluted EPS impact tied to integration expenses, intangible amortization expense, and loan and deposit fair value market amortization related to the Discover acquisition. COF YTD mountain Capital One YTD Shares were trading up about 4% in extended trading Tuesday night to around $226 per share. The stock’s record close is $229.74 set on Sept. 18. Bottom line The biggest takeaway from the quarter was better-than-expected credit performance. Credit has become a hot topic in the market lately due to the notable collapses of auto parts manufacturer First Brands Group and the subprime auto lender Tricolor Holdings. A few regional banks also recently flagged bad loans tied to possible fraud. Since Capital One has a large exposure to the subprime market, some investors weren’t quite sure how its loans were holding up. The subprime market is usually the first to feel the pain of an economic slowdown. That’s why it was so important to see Capital One once again report strong credit metrics, with better-than-expected net charge-offs and provisions for credit losses, resulting in an allowance release of $760 million and a boost to earnings per share by almost $1. Provisions for credit losses are funds that Capital One sets aside to cover potential loan defaults – the higher the provisions, the worse the sign of credit quality. The $760 million reflected continued favorable credit performance but was partially offset by what the company called “emerging economic uncertainties.” Why we own it Capital One’s acquisition of Discover is a transformative deal with significant strategic advantages and financial benefits. There are also several billion dollars worth of expenses and network synergies that should make this deal highly accretive to earnings per share. Lastly, the acquisition strengthens Capital One’s balance sheet, allowing for aggressive share repurchases in the future. Competitors : American Express , MasterCard , Visa Most recent buy : July 31, 2025 Initiated : March 6, 2025 Beyond the scrutiny of credit metrics, the other focus of Tuesday night’s post-earnings conference call was $35 billion acquisition of Discover and its integration. The company is still in its early stages, but the initial results are encouraging, with all synergies on track. “As a result of years of strategic preparation, we have a wealth of opportunities today that put us in an advantageous position to grow and win in the marketplace as it continues to change dramatically,” CEO Richard Fairbank said on the earnings call. “To capitalize on these opportunities at this special moment, we need to make significant and sustained investments.” He added, “Our acquisition of Discover enhances and accelerates some of these opportunities, and, of course, brings new opportunities as well.” Our thesis remains that the Discover acquisition will improve Capital One’s earnings power and help re-rate its price-to-earnings multiple as the business model evolves to more closely resemble American Express. By the way, we can check one box off our thesis after the company revealed a huge new share repurchase authorization. We’re reiterating our buy-equivalent 1 rating and price target of $250. Deal outlook On the earnings call, Capital One provided a positive update on the progress of the Discover integration. Consistent with what we learned last quarter, the company expects integration costs to be “somewhat higher” than its previously announced target of $2.8 billion. On the synergy side, Capital One said it’s on track to hit its target of $2.5 billion of net synergies, which is made up of cost savings and revenue synergies generated by moving its debit business and some of its credit business onto the Discover network. Fairbank said the process of moving its Capital One debit business off external networks like Mastercard’s and onto the Discover network is “going well” and will largely be completed in early 2026. Revenue synergies are expected to ramp up in the fourth quarter and in early 2026. From an operating expense standpoint, Fairbank said they are making “good progress.” In terms of long-term strategy, Fairbank wants to win at the top of the credit card market and pursue heavy spenders. He understands that winning in this market requires a lot of sustained investment to create a great product and give customers access to great experiences and exclusive investments. In a nod to JPMorgan’s Chase Reserve and American Express’ Platinum card, Fairbank noted that its biggest competitors in the premium space have “hugely stepped up their levels of investment,” and he wants to do the same. Commentary Capital One’s domestic card portfolio saw its net charge-off rate decline 64 basis points year over year to 3.89%. Net charge-offs refer to the amount of debt a bank has written off as uncollectible, minus any recoveries – a decline is a good thing. “Our charge-off rate has been improving on a seasonally adjusted basis throughout 2025 following the trend of improving delinquencies that started in late 2024 and supported by strong recoveries,” Fairbank explained. Within its consumer banking business, auto net charge-offs were 4.99%, down 62 basis points from the prior year. The auto results should ease fears around Capital One, considering the First Brands and Tricolor bankruptcies. “There’s been a lot of noise in the subprime auto space pointing to rising delinquency rates. Our own performance in subprime auto has remained stable through this time,” Fairbank said. Capital One’s subprime auto exposure is holding up better than peers thanks to a decision it made a few years ago to scale back its auto lending. Fairbank said the company anticipated inflated credit scores, normalizing credit, and declining vehicle values. As for buybacks, we learned during conference season that management stepped up the repurchase program, and they did not disappoint. The company repurchased 4.6 million shares for $1 billion in the third quarter, a sizeable increase from the $150 million worth of stock repurchased in the second quarter. The company also announced a new authorization of up to $16 billion, representing nearly 12% of the company’s current market cap. The company didn’t provide explicit guidance on how quickly it will move through this program, but CFO Andrew Young said that it’s “reasonable to assume that we’ll be picking up the pace of share repurchases from here.” Capital One also boosted the quarterly dividend by 20 cents per share to 80 cents. This bump increases the dividend yield to about 1.5% from $1.11%. (Jim Cramer’s Charitable Trust is long COF. See here for a full list of the stocks.) As a subscriber to the CNBC Investing Club with Jim Cramer, you will receive a trade alert before Jim makes a trade. Jim waits 45 minutes after sending a trade alert before buying or selling a stock in his charitable trust’s portfolio. If Jim has talked about a stock on CNBC TV, he waits 72 hours after issuing the trade alert before executing the trade. THE ABOVE INVESTING CLUB INFORMATION IS SUBJECT TO OUR TERMS AND CONDITIONS AND PRIVACY POLICY , TOGETHER WITH OUR DISCLAIMER . NO FIDUCIARY OBLIGATION OR DUTY EXISTS, OR IS CREATED, BY VIRTUE OF YOUR RECEIPT OF ANY INFORMATION PROVIDED IN CONNECTION WITH THE INVESTING CLUB. NO SPECIFIC OUTCOME OR PROFIT IS GUARANTEED.
Blog
-
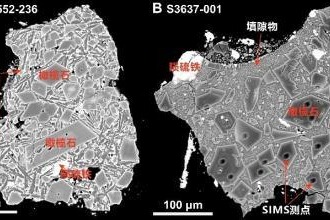
Soil sample study offers clues on lunar water evolution
Chinese scientists studying a 2-gram lunar soil sample from the Chang’e 6 mission have identified rare CI chondrite impact residues, providing new insights into mass transfer in the inner solar system and offering new…
Continue Reading
-
Inside Alana Hadid’s backyard wedding in LA—where sisters Bella and Gigi were barefoot bridesmaids – Vogue Australia
- Inside Alana Hadid’s backyard wedding in LA—where sisters Bella and Gigi were barefoot bridesmaids Vogue Australia
- Inside Alana Hadid’s Backyard L.A. Wedding—Where Sisters Bella and Gigi Were Barefoot Bridesmaids Vogue
- Alana Hadid gets…
Continue Reading
-

PADCEV™ (enfortumab vedotin-ejfv) Plus KEYTRUDA® (pembrolizumab) sBLA Granted FDA Priority Review for Treatment of Certain Patients with Muscle-Invasive Bladder Cancer
Results from the pivotal EV-303 trial demonstrated that, when used before and after surgery, the combination reduced the risk of recurrence, progression or death by 60% and the risk of death by 50% in cisplatin-ineligible patients with muscle-invasive bladder cancer
If approved, PADCEV plus KEYTRUDA could fundamentally change the treatment approach for patients with this disease
TOKYO, Oct. 22, 2025 /PRNewswire/ — Astellas Pharma Inc. (TSE: 4503, President and CEO: Naoki Okamura, “Astellas”) today announced that the U.S. Food and Drug Administration (FDA) accepted for priority review a supplemental Biologics License Application (sBLA) for PADCEV™ (enfortumab vedotin-ejfv) in combination with KEYTRUDA® (pembrolizumab) as a neoadjuvant treatment (before surgery) and then continued after radical cystectomy as adjuvant treatment (after surgery) for patients with muscle-invasive bladder cancer (MIBC) who are ineligible for cisplatin-containing chemotherapy.
Under the Prescription Drug User Fee Act (PDUFA), the FDA has set a target action date of April 7, 2026.
The sBLA submission was based on results from the pivotal Phase 3 EV-303 clinical trial (also known as KEYNOTE-905) evaluating PADCEV, a Nectin-4 directed antibody-drug conjugate, in combination with KEYTRUDA, a PD-1 inhibitor, as neoadjuvant and adjuvant treatment versus surgery alone, the current standard of care. Detailed results from EV-303, which showed the combination reduced the risk of recurrence, progression or death by 60% and the risk of death by 50% compared to surgery alone, were presented at the 2025 European Society of Medical Oncology (ESMO) Congress. The safety results in EV-303 were consistent with those previously reported for this combination, and no new safety signals were identified.
About the EV-303/KEYNOTE-905 Trial
The EV-303 trial (also known as KEYNOTE-905) is an ongoing, open-label, randomized, three-arm, controlled, Phase 3 study evaluating neoadjuvant and adjuvant PADCEV in combination with KEYTRUDA or neoadjuvant and adjuvant KEYTRUDA versus surgery alone in patients with MIBC who are either not eligible for or declined cisplatin-based chemotherapy. Patients were randomized to receive either neoadjuvant and adjuvant KEYTRUDA (arm A), surgery alone (arm B) or neoadjuvant and adjuvant PADCEV in combination with KEYTRUDA (arm C).1The primary endpoint of this trial is event-free survival (EFS) between arm C and arm B, defined as the time from randomization to the first occurrence of any of the following events: progression of disease that precludes radical cystectomy (RC) surgery or failure to undergo RC surgery in participants with residual disease, gross residual disease left behind at the time of surgery, local or distant recurrence as assessed by imaging and/or biopsy or death due to any cause. Key secondary endpoints include overall survival (OS) and pathologic complete response (pCR) rate between arm C and arm B, as well as EFS, OS and pCR rate between arm A and arm B.1
For more information on the global EV-303 trial, go to clinicaltrials.gov.
About Muscle-Invasive Bladder Cancer
Bladder cancer is the ninth most common cancer worldwide, diagnosed in more than 614,000 people each year globally, including an estimated 85,000 people in the U.S.2,3 MIBC represents approximately 30% of all bladder cancer cases.4 The standard treatment for patients with MIBC is neoadjuvant cisplatin-based chemotherapy followed by surgery, which has been shown to prolong survival.5 However, up to half of patients with MIBC are not eligible to receive cisplatin and face limited treatment options, typically undergoing surgery without any systemic treatment.6About PADCEV™ (enfortumab vedotin)
PADCEV™ (enfortumab vedotin) is a first-in-class antibody-drug conjugate (ADC) that is directed against Nectin-4, a protein located on the surface of cells and highly expressed in bladder cancer.7 Nonclinical data suggest the anticancer activity of PADCEV is due to its binding to Nectin-4-expressing cells, followed by the internalization and release of the anti-tumor agent monomethyl auristatin E (MMAE) into the cell, which result in the cell not reproducing (cell cycle arrest) and in programmed cell death (apoptosis).8PADCEV plus KEYTRUDA is approved for the treatment of adult patients with locally advanced or metastatic urothelial cancer (la/mUC) regardless of cisplatin eligibility in the United States, the European Union, Japan and a number of other countries around the world. PADCEV is also approved as a single agent for the treatment of adult patients with la/mUC who have previously received a PD-1/PD-L1 inhibitor and platinum-containing chemotherapy or are ineligible for cisplatin-containing chemotherapy and have previously received one or more prior lines of therapy.8
PADCEV (enfortumab vedotin-ejfv) U.S. Indication & Important Safety Information
BOXED WARNING: SERIOUS SKIN REACTIONS
- PADCEV can cause severe and fatal cutaneous adverse reactions including Stevens-Johnson syndrome (SJS) and Toxic Epidermal Necrolysis (TEN), which occurred predominantly during the first cycle of treatment, but may occur later.
- Closely monitor patients for skin reactions.
- Immediately withhold PADCEV and consider referral for specialized care for suspected SJS or TEN or severe skin reactions.
- Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions.
Indication
PADCEV™, in combination with pembrolizumab, is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC).PADCEV, as a single agent, is indicated for the treatment of adult patients with locally advanced or mUC who:
- have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and platinum-containing chemotherapy, or
- are ineligible for cisplatin-containing chemotherapy and have previously received one or more prior lines of therapy.
PADCEV Important Safety Information
Warnings and Precautions
Skin reactions Severe cutaneous adverse reactions, including fatal cases of SJS or TEN occurred in patients treated with PADCEV. SJS and TEN occurred predominantly during the first cycle of treatment but may occur later. Skin reactions occurred in 70% (all grades) of the 564 patients treated with PADCEV in combination with pembrolizumab in clinical trials. When PADCEV was given in combination with pembrolizumab, the incidence of skin reactions, including severe events, occurred at a higher rate compared to PADCEV as a single agent. The majority of the skin reactions that occurred with combination therapy included maculo-papular rash, macular rash and papular rash. Grade 3-4 skin reactions occurred in 17% of patients (Grade 3: 16%, Grade 4: 1%), including maculo-papular rash, bullous dermatitis, dermatitis, exfoliative dermatitis, pemphigoid, rash, erythematous rash, macular rash, and papular rash. A fatal reaction of bullous dermatitis occurred in one patient (0.2%). The median time to onset of severe skin reactions was 1.7 months (range: 0.1 to 17.2 months). Skin reactions led to discontinuation of PADCEV in 6% of patients.
Skin reactions occurred in 58% (all grades) of the 720 patients treated with PADCEV as a single agent in clinical trials. Twenty-three percent (23%) of patients had maculo-papular rash and 34% had pruritus. Grade 3-4 skin reactions occurred in 14% of patients, including maculo-papular rash, erythematous rash, rash or drug eruption, symmetrical drug-related intertriginous and flexural exanthema (SDRIFE), bullous dermatitis, exfoliative dermatitis, and palmar-plantar erythrodysesthesia. The median time to onset of severe skin reactions was 0.6 months (range: 0.1 to 8 months). Among patients experiencing a skin reaction leading to dose interruption who then restarted PADCEV (n=75), 24% of patients restarting at the same dose and 24% of patients restarting at a reduced dose experienced recurrent severe skin reactions. Skin reactions led to discontinuation of PADCEV in 3.1% of patients.
Monitor patients closely throughout treatment for skin reactions. Consider topical corticosteroids and antihistamines, as clinically indicated. For persistent or recurrent Grade 2 skin reactions, consider withholding PADCEV until Grade ≤1. Withhold PADCEV and refer for specialized care for suspected SJS, TEN or for Grade 3 skin reactions. Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions.
Hyperglycemia and diabetic ketoacidosis (DKA), including fatal events, occurred in patients with and without pre-existing diabetes mellitus, treated with PADCEV. Patients with baseline hemoglobin A1C ≥8% were excluded from clinical trials. In clinical trials of PADCEV as a single agent, 17% of the 720 patients treated with PADCEV developed hyperglycemia of any grade; 7% of patients developed Grade 3-4 hyperglycemia (Grade 3: 6.5%, Grade 4: 0.6%). Fatal events of hyperglycemia and DKA occurred in one patient each (0.1%). The incidence of Grade 3-4 hyperglycemia increased consistently in patients with higher body mass index and in patients with higher baseline A1C. The median time to onset of hyperglycemia was 0.5 months (range: 0 to 20 months). Hyperglycemia led to discontinuation of PADCEV in 0.7% of patients. Five percent (5%) of patients required initiation of insulin therapy for treatment of hyperglycemia. Of the patients who initiated insulin therapy for treatment of hyperglycemia, 66% (23/35) discontinued insulin at the time of last evaluation. Closely monitor blood glucose levels in patients with, or at risk for, diabetes mellitus or hyperglycemia. If blood glucose is elevated (>250 mg/dL), withhold PADCEV.
Pneumonitis/Interstitial Lung Disease (ILD) Severe, life-threatening or fatal pneumonitis/ILD occurred in patients treated with PADCEV. When PADCEV was given in combination with pembrolizumab, 10% of the 564 patients treated with combination therapy had pneumonitis/ILD of any grade and 4% had Grade 3-4. A fatal event of pneumonitis/ILD occurred in two patients (0.4%). The incidence of pneumonitis/ILD, including severe events, occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of any grade pneumonitis/ILD was 4 months (range: 0.3 to 26 months).
In clinical trials of PADCEV as a single agent, 3% of the 720 patients treated with PADCEV had pneumonitis/ILD of any grade and 0.8% had Grade 3-4. The median time to onset of any grade pneumonitis/ILD was 2.9 months (range: 0.6 to 6 months).
Monitor patients for signs and symptoms indicative of pneumonitis/ILD such as hypoxia, cough, dyspnea or interstitial infiltrates on radiologic exams. Evaluate and exclude infectious, neoplastic and other causes for such signs and symptoms through appropriate investigations. Withhold PADCEV for patients who develop Grade 2 pneumonitis/ILD and consider dose reduction. Permanently discontinue PADCEV in all patients with Grade 3 or 4 pneumonitis/ILD.
Peripheral neuropathy (PN) When PADCEV was given in combination with pembrolizumab, 67% of the 564 patients treated with combination therapy had PN of any grade, 36% had Grade 2 neuropathy, and 7% had Grade 3 neuropathy. The incidence of PN occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of Grade ≥2 PN was 6 months (range: 0.3 to 25 months).
PN occurred in 53% of the 720 patients treated with PADCEV as a single agent in clinical trials including 38% with sensory neuropathy, 8% with muscular weakness and 7% with motor neuropathy. Thirty percent of patients experienced Grade 2 reactions and 5% experienced Grade 3-4 reactions. PN occurred in patients treated with PADCEV with or without preexisting PN. The median time to onset of Grade ≥2 PN was 4.9 months (range: 0.1 to 20 months). Neuropathy led to treatment discontinuation in 6% of patients.
Monitor patients for symptoms of new or worsening PN and consider dose interruption or dose reduction of PADCEV when PN occurs. Permanently discontinue PADCEV in patients who develop Grade ≥3 PN.
Ocular disorders were reported in 40% of the 384 patients treated with PADCEV as a single agent in clinical trials in which ophthalmologic exams were scheduled. The majority of these events involved the cornea and included events associated with dry eye such as keratitis, blurred vision, increased lacrimation, conjunctivitis, limbal stem cell deficiency, and keratopathy. Dry eye symptoms occurred in 30% of patients, and blurred vision occurred in 10% of patients, during treatment with PADCEV. The median time to onset to symptomatic ocular disorder was 1.7 months (range: 0 to 30.6 months). Monitor patients for ocular disorders. Consider artificial tears for prophylaxis of dry eyes and ophthalmologic evaluation if ocular symptoms occur or do not resolve. Consider treatment with ophthalmic topical steroids, if indicated after an ophthalmic exam. Consider dose interruption or dose reduction of PADCEV for symptomatic ocular disorders.
Infusion site extravasation Skin and soft tissue reactions secondary to extravasation have been observed after administration of PADCEV. Of the 720 patients treated with PADCEV as a single agent in clinical trials, 1% of patients experienced skin and soft tissue reactions, including 0.3% who experienced Grade 3-4 reactions. Reactions may be delayed. Erythema, swelling, increased temperature, and pain worsened until 2-7 days after extravasation and resolved within 1-4 weeks of peak. Two patients (0.3%) developed extravasation reactions with secondary cellulitis, bullae, or exfoliation. Ensure adequate venous access prior to starting PADCEV and monitor for possible extravasation during administration. If extravasation occurs, stop the infusion and monitor for adverse reactions.
Embryo-fetal toxicity PADCEV can cause fetal harm when administered to a pregnant woman. Advise patients of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during PADCEV treatment and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with PADCEV and for 4 months after the last dose.
ADVERSE REACTIONS
Most common adverse reactions, including laboratory abnormalities (≥20%) (PADCEV in combination with pembrolizumab)
Increased aspartate aminotransferase (AST), increased creatinine, rash, increased glucose, PN, increased lipase, decreased lymphocytes, increased alanine aminotransferase (ALT), decreased hemoglobin, fatigue, decreased sodium, decreased phosphate, decreased albumin, pruritus, diarrhea, alopecia, decreased weight, decreased appetite, increased urate, decreased neutrophils, decreased potassium, dry eye, nausea, constipation, increased potassium, dysgeusia, urinary tract infection and decreased platelets.Most common adverse reactions, including laboratory abnormalities (≥20%) (PADCEV monotherapy)
Increased glucose, increased AST, decreased lymphocytes, increased creatinine, rash, fatigue, PN, decreased albumin, decreased hemoglobin, alopecia, decreased appetite, decreased neutrophils, decreased sodium, increased ALT, decreased phosphate, diarrhea, nausea, pruritus, increased urate, dry eye, dysgeusia, constipation, increased lipase, decreased weight, decreased platelets, abdominal pain, dry skin.EV-302 Study: 440 patients with previously untreated la/mUC (PADCEV in combination with pembrolizumab)
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab. The most common serious adverse reactions (≥2%) were rash (6%), acute kidney injury (5%), pneumonitis/ILD (4.5%), urinary tract infection (3.6%), diarrhea (3.2%), pneumonia (2.3%), pyrexia (2%), and hyperglycemia (2%). Fatal adverse reactions occurred in 3.9% of patients treated with PADCEV in combination with pembrolizumab including acute respiratory failure (0.7%), pneumonia (0.5%), and pneumonitis/ILD (0.2%).Adverse reactions leading to discontinuation of PADCEV occurred in 35% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were PN (15%), rash (4.1%) and pneumonitis/ILD (2.3%). Adverse reactions leading to dose interruption of PADCEV occurred in 73% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were PN (22%), rash (16%), COVID19 (10%), diarrhea (5%), pneumonitis/ILD (4.8%), fatigue (3.9%), hyperglycemia (3.6%), increased ALT (3%) and pruritus (2.5%). Adverse reactions leading to dose reduction of PADCEV occurred in 42% of patients. The most common adverse reactions (≥2%) leading to dose reduction of PADCEV were rash (16%), PN (13%) and fatigue (2.7%).
EV-103 Study: 121 patients with previously untreated la/mUC who were not eligible for cisplatin-containing chemotherapy (PADCEV in combination with pembrolizumab)
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab; the most common (≥2%) were acute kidney injury (7%), urinary tract infection (7%), urosepsis (5%), sepsis (3.3%), pneumonia (3.3%), hematuria (3.3%), pneumonitis/ILD (3.3%), urinary retention (2.5%), diarrhea (2.5%), myasthenia gravis (2.5%), myositis (2.5%), anemia (2.5%), and hypotension (2.5%). Fatal adverse reactions occurred in 5% of patients treated with PADCEV in combination with pembrolizumab, including sepsis (1.6%), bullous dermatitis (0.8%), myasthenia gravis (0.8%), and pneumonitis/ILD (0.8%). Adverse reactions leading to discontinuation of PADCEV occurred in 36% of patients; the most common (≥2%) were PN (20%) and rash (6%). Adverse reactions leading to dose interruption of PADCEV occurred in 69% of patients; the most common (≥2%) were PN (18%), rash (12%), increased lipase (6%), pneumonitis/ILD (6%), diarrhea (4.1%), acute kidney injury (3.3%), increased ALT (3.3%), fatigue (3.3%), neutropenia (3.3%), urinary tract infection (3.3%), increased amylase (2.5%), anemia (2.5%), COVID19 (2.5%), hyperglycemia (2.5%), and hypotension (2.5%). Adverse reactions leading to dose reduction of PADCEV occurred in 45% of patients; the most common (≥2%) were PN (17%), rash (12%), fatigue (5%), neutropenia (5%), and diarrhea (4.1%).EV-301 Study: 296 patients previously treated with a PD-1/L1 inhibitor and platinum-based chemotherapy (PADCEV monotherapy)
Serious adverse reactions occurred in 47% of patients treated with PADCEV; the most common (≥2%) were urinary tract infection, acute kidney injury (7% each), and pneumonia (5%). Fatal adverse reactions occurred in 3% of patients, including multiorgan dysfunction (1%), hepatic dysfunction, septic shock, hyperglycemia, pneumonitis/ILD, and pelvic abscess (0.3% each). Adverse reactions leading to discontinuation occurred in 17% of patients; the most common (≥2%) were PN (5%) and rash (4%). Adverse reactions leading to dose interruption occurred in 61% of patients; the most common (≥4%) were PN (23%), rash (11%), and fatigue (9%). Adverse reactions leading to dose reduction occurred in 34% of patients; the most common (≥2%) were PN (10%), rash (8%), decreased appetite, and fatigue (3% each).EV-201, Cohort 2 Study: 89 patients previously treated with a PD-1/L1 inhibitor and not eligible for cisplatin-based chemotherapy (PADCEV monotherapy)
Serious adverse reactions occurred in 39% of patients treated with PADCEV; the most common (≥3%) were pneumonia, sepsis, and diarrhea (5% each). Fatal adverse reactions occurred in 8% of patients, including acute kidney injury (2.2%), metabolic acidosis, sepsis, multiorgan dysfunction, pneumonia, and pneumonitis/ILD (1.1% each). Adverse reactions leading to discontinuation occurred in 20% of patients; the most common (≥2%) was PN (7%). Adverse reactions leading to dose interruption occurred in 60% of patients; the most common (≥3%) were PN (19%), rash (9%), fatigue (8%), diarrhea (5%), increased AST, and hyperglycemia (3% each). Adverse reactions leading to dose reduction occurred in 49% of patients; the most common (≥3%) were PN (19%), rash (11%), and fatigue (7%).DRUG INTERACTIONS
Effects of other drugs on PADCEV (Dual P-gp and Strong CYP3A4 Inhibitors)
Concomitant use with dual P-gp and strong CYP3A4 inhibitors may increase unconjugated monomethyl auristatin E exposure, which may increase the incidence or severity of PADCEV toxicities. Closely monitor patients for signs of toxicity when PADCEV is given concomitantly with dual P-gp and strong CYP3A4 inhibitors.SPECIFIC POPULATIONS
Lactation Advise lactating women not to breastfeed during treatment with PADCEV and for 3 weeks after the last dose.Hepatic impairment Avoid the use of PADCEV in patients with moderate or severe hepatic impairment.
For more information, please see the U.S. full Prescribing Information including BOXED WARNING for PADCEV here .
About Astellas
Astellas is a global life sciences company committed to turning innovative science into VALUE for patients. We provide transformative therapies in disease areas that include oncology, ophthalmology, urology, immunology and women’s health. Through our research and development programs, we are pioneering new healthcare solutions for diseases with high unmet medical need. Learn more at www.astellas.com.About the Pfizer, Astellas and Merck Collaboration
Seagen and Astellas entered a clinical collaboration agreement with Merck to evaluate the combination of Seagen’s and Astellas’ PADCEV™ (enfortumab vedotin-ejfv) and Merck’s KEYTRUDA® (pembrolizumab) in patients with previously untreated metastatic urothelial cancer. Pfizer Inc. successfully completed its acquisition of Seagen on December 14, 2023. KEYTRUDA is a registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (known as MSD outside of the United States and Canada).Cautionary Notes
In this press release, statements made with respect to current plans, estimates, strategies and beliefs and other statements that are not historical facts are forward-looking statements about the future performance of Astellas. These statements are based on management’s current assumptions and beliefs in light of the information currently available to it and involve known and unknown risks and uncertainties. A number of factors could cause actual results to differ materially from those discussed in the forward-looking statements. Such factors include, but are not limited to: (i) changes in general economic conditions and in laws and regulations, relating to pharmaceutical markets, (ii) currency exchange rate fluctuations, (iii) delays in new product launches, (iv) the inability of Astellas to market existing and new products effectively, (v) the inability of Astellas to continue to effectively research and develop products accepted by customers in highly competitive markets, and (vi) infringements of Astellas ‘ intellectual property rights by third parties. Information about pharmaceutical products (including products currently in development) which is included in this press release is not intended to constitute an advertisement or medical advice.References:
1 National Institute of Health. National Library of Medicine. Perioperative Pembrolizumab (MK-3475) Plus Cystectomy or Perioperative Pembrolizumab Plus Enfortumab Vedotin Plus Cystectomy Versus Cystectomy Alone in Participants Who Are Cisplatin-ineligible or Decline Cisplatin With Muscle-invasive Bladder Cancer (MK-3475-905/ KEYNOTE-905/ EV-303. ClinicalTrials.gov identifier: NCT03924895. Published July 24, 2019. Updated June 17, 2025. Accessed June 23, 2025. Available at: https://clinicaltrials.gov/study/NCT03924895?term=AREA%5BBasicSearch%5D(myosarcoma)&rank=3
2 World Bladder Cancer Patient Coalition. GLOBOCAN 2022: Bladder cancer 9th most common worldwide. Accessed June 23, 2025. Available at: https://worldbladdercancer.org/news_events/globocan-2022-bladder-cancer-is-the-9th-most-commonly-diagnosed-worldwide/
3 American Cancer Society. Cancer Facts & Figures 2025. Accessed September 23, 2025. Available at: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2025-cancer-facts-figures.html
4 Bladder Cancer Awareness Network. What is Muscle Invasive Bladder Cancer? Accessed June 23, 2025. Available at: https://bcan.org/what-is-muscle-invasive-bladder-cancer/#:~:text=When%20tumors%20grow%20into%20or,Virginia%20Health%20System%20explain%20MIBC.
5 Funt SA, Rosenberg JE. Systemic, perioperative management of muscle-invasive bladder cancer and future horizons. Nat Rev Clin Oncol. 2017 Apr;14(4):221-234. doi: 10.1038/nrclinonc.2016.188. Epub 2016 Nov 22. PMID: 27874062; PMCID: PMC6054138.
6 Esteban-Villarrubia J, Torres-Jiménez J, Bueno-Bravo C, García-Mondaray R, Subiela JD, Gajate P. Current and Future Landscape of Perioperative Treatment for Muscle-Invasive Bladder Cancer. Cancers (Basel). 2023 Jan 17;15(3):566. doi: 10.3390/cancers15030566. PMID: 36765525; PMCID: PMC9913718.
7 Challita-Eid PM, Satpayev D, Yang P, et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res 2016;76(10):3003-13.
8 PADCEV [package insert]. Northbrook, IL: Astellas Pharma US, Inc.SOURCE Astellas Pharma Inc.
Continue Reading
-
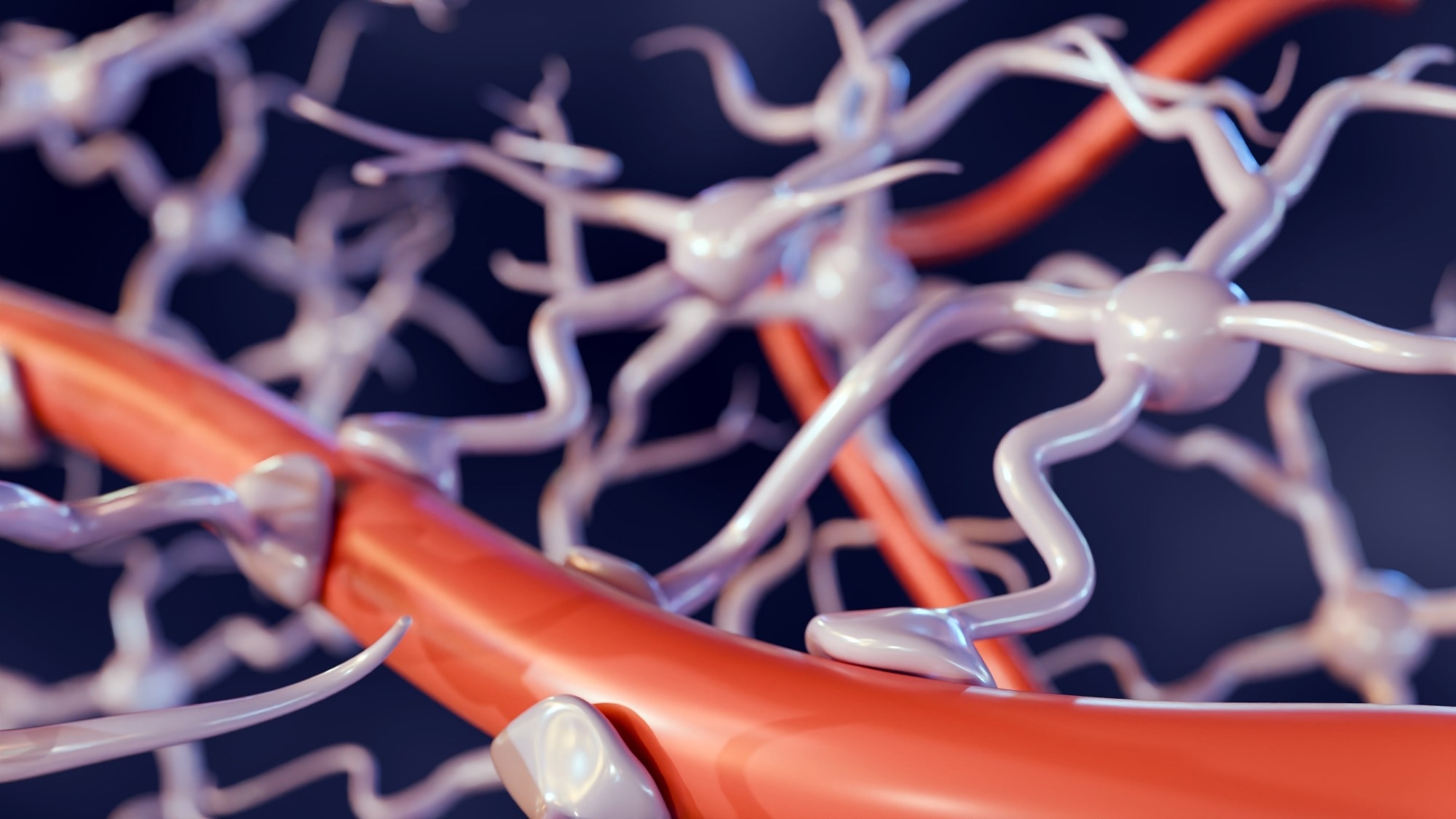
New insights into how the blood-brain barrier safeguards the brain
By decoding the blood-brain barrier’s intricate structure and function, researchers are paving the way for new therapies that can safely breach the brain’s toughest defense.
Primer: The blood–brain barrier. Image Credit: Love…
Continue Reading
-
Faraday Future Announces its Official Seven-Day Countdown and Livestream Details for its FX Super One MPV Middle East Launch Event, to be Held in Dubai, UAE, on October 28, 2025 – Faraday Future
- Faraday Future Announces its Official Seven-Day Countdown and Livestream Details for its FX Super One MPV Middle East Launch Event, to be Held in Dubai, UAE, on October 28, 2025 Faraday Future
- Oct. 28 UAE launch — FX Super One enters pilot build as parts ship; Faraday Future targets year‑end off‑line Stock Titan
- Faraday Future Founder and Co-CEO YT Jia Shares Weekly Investor Update: The Whole FX Team is … Bluefield Daily Telegraph
- Faraday Future Plans FX Super One Launch in Dubai TipRanks
- Faraday Future Founder and Co-CEO YT Jia Shares Weekly GlobeNewswire
Continue Reading
-

Knicks will be without Josh Hart, Mitchell Robinson in season opener
Mitchell Robinson (right) had ankle surgery before last season and was limited to 17 games after he returned.
NEW YORK (AP) — New York Knicks coach Mike Brown will have to wait to show whether Josh Hart or Mitchell Robinson will start for him….
Continue Reading
-

Champions League: An eventful evening as 43 goals scored, five red cards given, six penalties awarded
There were 43 goals scored, five red cards handed out and six penalties awarded – of which five were converted.
It was a relentless evening of Champions League football on Tuesday.
Last season’s winners Paris St-Germain hit seven past Leverkusen,…
Continue Reading
-
Just a moment…
Just a moment… This request seems a bit unusual, so we need to confirm that you’re human. Please press and hold the button until it turns completely green. Thank you for your cooperation!
Continue Reading
-
Just a moment…
Just a moment… This request seems a bit unusual, so we need to confirm that you’re human. Please press and hold the button until it turns completely green. Thank you for your cooperation!
Continue Reading