DNA viruses in the detected samples
By assembling and analyzing the metagenomic sequencing data, this study only discovered two virus contig sequences, tig000081 and tig000315, which possessed the characteristics of Caulimoviridae viruses. Therefore, the two novel Caulimoviridae viruses, tig000081 (7,546 nt) and tig000315 (7,585 nt) are tentatively named “Lancifolium Caulimovirus A” (LCaA) and “Lancifolium Caulimovirus B” (LCaB), respectively.
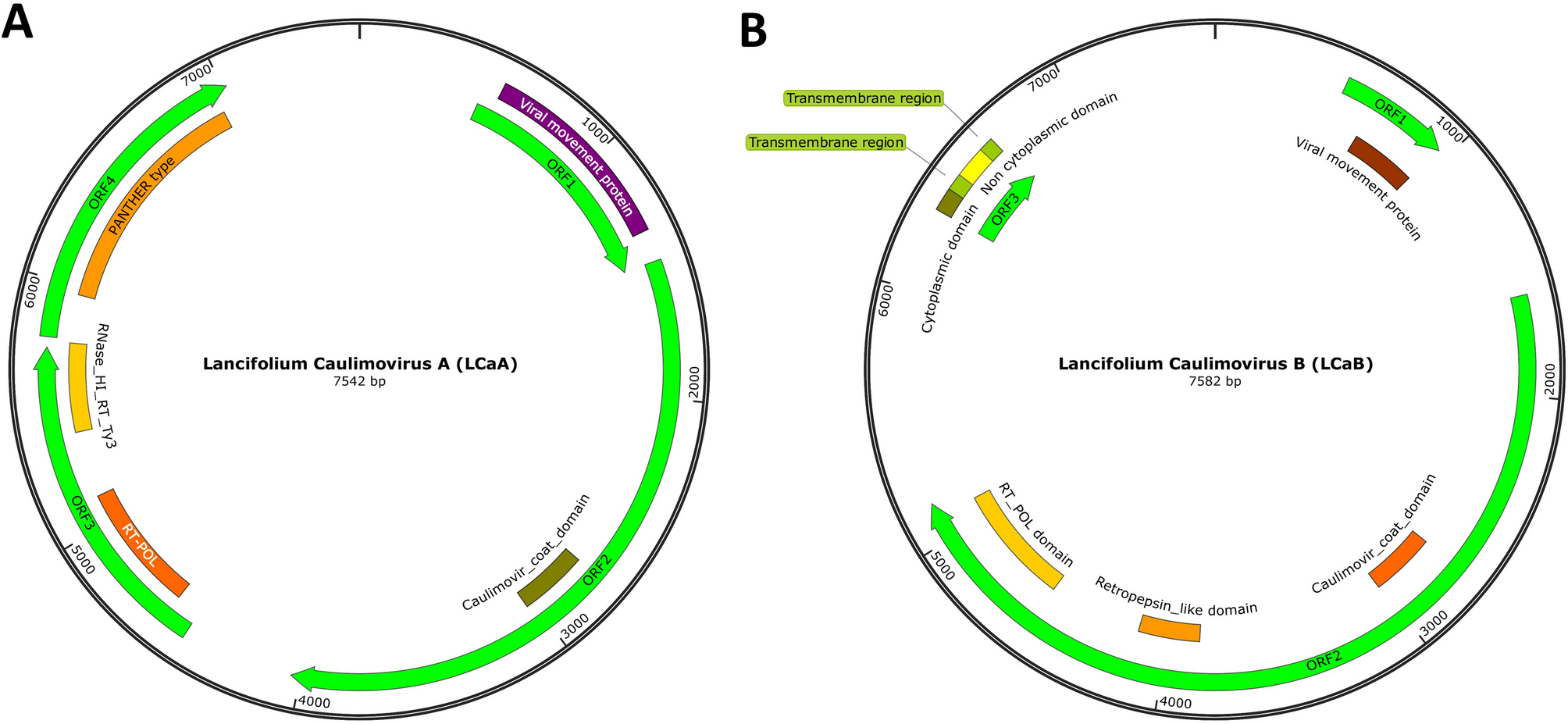
To validate the accuracy of the two reference contig sequences, overlapping fragments from LCaA and LCaB were cloned, sequenced, and assembled to obtain full-length genomic sequences. Final genome lengths were determined as 7,542 nt for LCaA and 7,582 nt for LCaB, with complete sequences deposited in the China National GeneBank Database (CNGB) under accession number sub064865. The open reading frame (ORF) prediction using ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) and conserved domain analysis via InterPro (https://www.ebi.ac.uk/interpro/) revealed distinct genomic architectures of the two viruses. For LCaA, it had four ORFs: ORF1 was 963 nt and encoded a viral movement protein (MP) (aa 23–277), which was necessary for initial cell-to-cell movement during the early stages of viral infection and predicted to be responsible for the intercellular transport of viral genomes in plant cells [29]. ORF2 was 2,571 nt and encoded an 856-amino acid protein containing one Caulimovir coat domain (amino acids 427–522), homologous to coat proteins in Cauliflower mosaic virus. ORF3 was 1,656 nt and encoded a viral replication and maturation polyprotein (amino acids 104–527). This polyprotein contained one RT POL domain (139–318) and one RNase HI RT Ty3 domain (412–535). The retroviral reverse transcriptase (prototypical RT) had three enzymatic activities: RNA-dependent DNA polymerase, ribonuclease H, and DNA-dependent DNA polymerase. These activities involve copying the plus-strand RNA genome to produce minus-strand DNA, removing the RNA template, and synthesizing plus-strand DNA using minus-strand DNA as a template [30]. ORF4 was 1,233 nt and encoded a hypothetical protein (amino acids 64–393) with an uncharacterized function. (Fig. 1A). Former research had reported that the CaMV genome always encodes six proteins, a cell-to-cell movement protein, two aphid transmission factors: a polyprotein precursor of proteinase, a precursor of the capsid proteins, the reverse transcriptase and ribonuclease H, and an inclusion body protein/translation transactivator [5, 31]. Our newly detected LCa virus processed most of the key proteins of the CaMV, and ORF2 containing one Caulimovir coat domain. Based on the description of the above-mentioned LCa virus characteristics, we believe that this virus belongs to the Cauliflower Mosaic Virus.
Circular representation map of the LCa genome. (A) LCaA genome. (B) LCaB genome
LCaB contained three ORFs. ORF1 encoded a viral movement protein (amino acids 47–145), which, like LCaA, was predicted to be a Caulimoviridae movement protein. ORF2 encoded a protein containing one caulimovir coat domain (amino acids 371–467), one retropepsin-like domain (amino acids 750–841), and one RT POL domain (amino acids 982–1,161). ORF3 encoded a protein with a cytoplasmic domain (amino acids 1–30), a transmembrane region (amino acids 31–50), a noncytoplasmic domain (amino acids 54–94), and another transmembrane region (amino acids 95–113) (Fig. 1B).
The genetic evolutionary relationship of the genus Caulimovirus
According to the International Committee on Taxonomy of Viruses (ICTV; https://ictv.global/), the genus Caulimovirus currently comprises 18 recognized species, including Cauliflower mosaic virus (CaMV) and eight other taxonomically validated members (Fig. 2). All the known genus Caulimovirus virus sequences were alignment with Mega 11 [32], and using maximum likelihood statistical method with 100 bootstrap replications to test the phylogeny. Then, the obtained tree file was up-loaded to the iTOL(https://itol.embl.de/) to beautify the graphics. Systematic phylogenetic analysis classified all reported Caulimovirus species into four well-supported monophyletic groups (Fig. 2). These species share a common evolutionary origin but subsequently diverged into four distinct lineages during key evolutionary transitions. Besides, the obtained phylogenetic tree showed that LCaA and LCaB share the closest evolutionary affinity, forming a distinct clade with Plant associated caulimovirus (PAC), Strawberry vein banding virus (SVBV), Pelargonium vein alating virus (PVA), and Malva yellow mosaic virus (MYMAV) (Fig. 2). That means within this branch, the genetic variation shared among species (the similarity of homologous gene sequences) is significantly higher than that between them and the species in other branches on the evolutionary tree. This indicates that they separated from each other for a shorter period during the evolutionary process and accumulated fewer genetic differences.
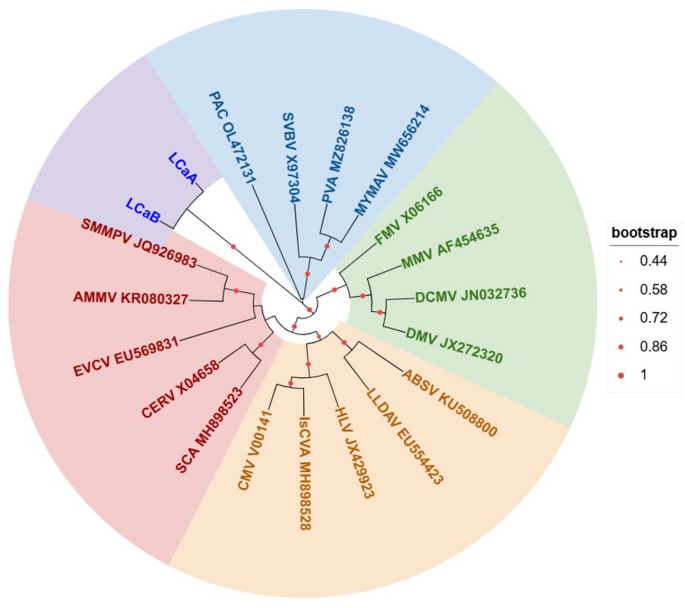
The genetic evolutionary relationship of the genus Caulimovirus. Mirabilis mosaic virus (MMV) AF454635, Carnation etched ring virus (CERV) CERV X04658, Figwort mosaic virus (FMV) FMV X06166, Cauliflower mosaic virus (CMV) V00141, Strawberry vein banding virus (SVBV) X97304, Eupatorium vein clearing virus (EVCV) EU569831, Lamium leaf distortion associated virus (LLDAV) EU554423, Horseradish latent virus (HLV) JX429923, Soybean mild mottle pararetrovirus (SMMPV) JQ926983, Dahlia mosaic virus (DMV) JX272320, Atractylodes mild mottle virus (AMMV) KR080327, Angelica bushy stunt virus (ABSV) KU508800, Metaplexis yellow mottle-associated virus (MYMAV) MW656214, Dahlia common mosaic virus (DCMV) JN032736, Isatis caulimovirus A (IsCVA) MH898528, Silene caulimovirus A (SCA) MH898523, Plant associated caulimovirus (PAC) OL472131, Pueraria virus A (PVA) MZ826138
Marker-based identification of LCa DNA viruses
The tiger lily plants suspected to be infected with the virus were selected, and their phynotypes were shown in Fig. 3A and B. Based on Sanger sequencing and assembly of virus fragments in the pooled samples, we observed natural genetic variations in the LCaA and LCaB viruses. We finally obtained seven LCaA virus isolates and six LCaB isolates, and their genome sequences were deposited in the China National GeneBank (CNGB; accession number sub064865) and National Center for Biotechnology Information (NCBI), which is also shown in Sub Table 2. The genome sequence similarities among LCaA virus isolates ranged from 99.59 to 99.73%, while those among LCaB isolates ranged from 98.51 to 98.91%. To identify these newly discovered LCa viruses, their genome sequences were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Based on conserved sequences shared by LCaA and LCaB, four marker primers (listed in Table 1) were specifically designed to detect LCa viruses. Following PCR amplification, Marker 1 produced amplicons of 519 bp (LCaA) and 513 bp (LCaB), whereas Marker 2 yielded 269 bp amplicons for both variants (Fig. 3D). Furthermore, using LCaA-specific conserved sequences, Marker 3 produced a 342 bp amplicon for LCaA. Combined detection using Markers 1, 2, and 3 enabled specific identification of LCaA viruses (Fig. 3D). Marker 4 generated a 203 bp amplicon using this approach. Using Markers 1, 2, and 4, LCaB viruses were reliably identified (Fig. 3D). Furthermore, the bulbs’ DNA of the marked samples in Shuanghe town were amplified using the four primer pairs. The results showed that the Shuanghe-1(SH-1) was infected with both LCaA and LCaB, the Shuanghe-2(SH-2) was infected with LCaA, the Shuanghe-3 (SH-3) was infected with LCaB, the Shuanghe-4(SH-4) not infected with LCa virus (Fig. 3E). The above experiments demonstrated that the four primer pairs enable efficient and specific identification of LCa viruses.

Virus infection symptoms and identification in tiger lilies. (A), (B), and (C) Phenotypic characteristics of tiger lilies suspected of virus infection. (D) Agarose gel electrophoresis showing virus identification markers. M: DL2000 marker; 1 and 2: PCR products amplified via marker 1 primers (LCa-SP-F2/LCa-SP-R2) from tiger lily’s total DNA infected with and not infected with LCa viruses respectively; 3 and 4: PCR products amplified via marker 2 primers (LCa-SP-F4/LCa-SP-R4) from tiger lily’s total DNA infected with and not infected with LCa viruses, respectively; 5 and 6: PCR products amplified via marker 3 primers (LCaA-SP-F/LCaA-SP-R) from tiger lily’s total DNA infected with and not infected with LCaA virus, respectively; 7 and 8: PCR products amplified via marker 4 primers (LCaB-SP-F/LCaB-SP-R) from tiger lily’s total DNA infected with and not infected with LCaB virus, respectively; 9, 11, and 14: PCR products amplified via marker 1 primers from tiger lily’s total DNA infected with LCa viruses; 10, 12, and 15: PCR products amplified via marker 2 primers from tiger lily’s total DNA infected with LCa viruses; 13: PCR products amplified via marker 3 primers from tiger lily’s total DNA infected with LCaA virus; 16: PCR products amplified via marker 4 primers from tiger lily’s total DNA infected with LCaB virus. (E) LCa virus identification in the tiger lily bulbs from Shuanghe (SH) town. M: DL2000 marker; 1, 5, 10, and 13: PCR products amplified via marker one primers; 2, 6, 11, and 14: PCR products amplified via marker two primers; 3, 7, 12, and 15: PCR products amplified via marker three primers; 4, 8, 13, and 16: PCR products amplified via marker four primers
At the same time, another forty tiger lily plants were randomly collected at each sample collecting point. Their DNA was extracted using the methods mentioned before. Furthermore, the extracted DNA was used as templates for PCR. Marker 1 and Marker 2 were used as the primers to detect the LCa virus in those randomly collected samples. The following identification experiments were performed as mentioned before. The results showed that the LCa virus incidence of the tiger lily in Shuanghe, Sancha, Caoba Xia, Zhengdian, and Dongshan was 10%, 1%, 2%, 2%, and 2%, respectively (Sub Fig. 1).
Viral abundance analysis of all detected viruses
We analyzed the DNA and RNA virus abundance based on the viral metagenomic and metatranscriptomic analysis. Only one Caulimoviridae family virus was detected using viral metagenomic sequencing data, and the above experiments demonstrated that this family Caulimoviridae virus is a newly found LCa virus. The viral abundance analysis using viral metatranscriptomic sequencing data, and the virus contigs information were provided in Sub Table 2. The viral abundance statistic data are shown in Sub Table 3. The result showed that order Sobelivirales account for 8%, Iris potyvirus A species account for 5%, Shallot yellow stripe virus species account for 4%, genus Potexvirus account for 8%, Lily symptomless virus species account for 7%, Cucumber mosaic virus species account for 3%, order Reovirales account for 0.1%, Lenarviricota phylum account for 0.005%, family Caulimoviridae account for 0.001%. Other viruses were unclassified, and nearly 64% unclassified viruses belong to the order Martellivirales. 2% of unclassified virus belong to the genus Carlavirus, and 2% belong to the order Tymovirales (Fig. 4).
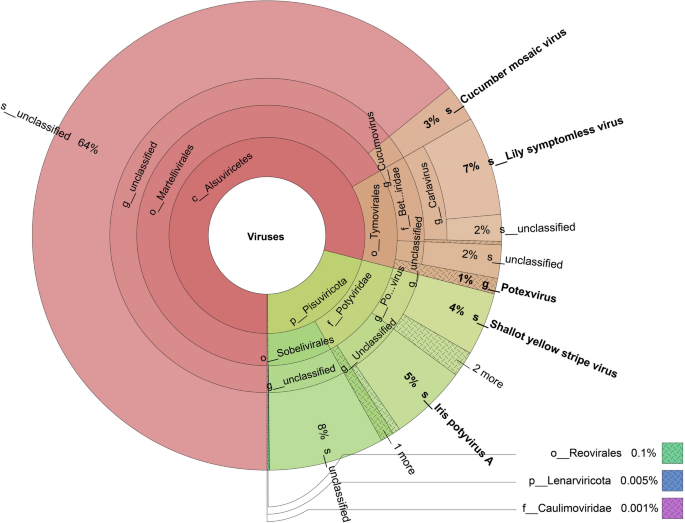
Viral abundance analysis based on the viral metatranscriptomic sequencing data