Ethics statement
This study complies with all relevant ethical regulations. TAMARISK specimens were obtained and sequenced with the approval of the institutional review boards (IRBs) of the Netherlands Cancer Institute (protocol CFMPB294) and the Dana-Farber Cancer Institute (DFCI) (protocol 12-049B). Approval to access clinical data from the DFCI was granted under protocols 17-000 and 11-104. All participants from both the TAMARISK and DFCI cohorts provided written informed consent, allowing their genomic and clinical data to be obtained and analyzed here. In accordance with the US Code of Federal Regulations, Title 45, Part 46, Section 104(d) (45 CFR §46.104(d)), the retrospective analysis of de-identified clinical data from Caris Life Sciences was deemed exempt by the IRB, which is the WIRB-Copernicus Group IRB (formerly known as WIRB). This exemption was granted because the data were fully de-identified and the research involved no intervention or interaction with human participants; therefore, informed patient consent was not required.
Tamoxifen-associated uterine cancer from the TAMARISK study
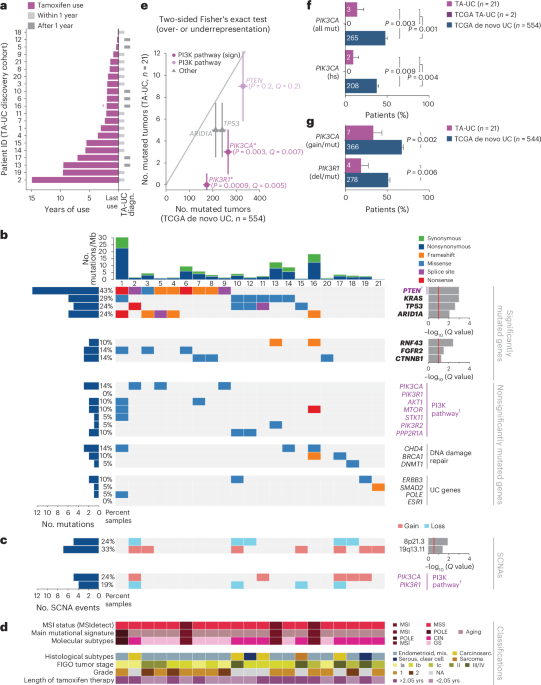
We analyzed 60 primary TA-UCs from the TAMARISK study28, diagnosed between 1983 and 2002, for which sufficient residual tissue for DNA extraction was available (Extended Data Fig. 1a and Supplementary Table 1). Of these, 21 samples and their matched normal counterparts underwent WES and constitute the discovery cohort. Another 39 TA-UC samples were subjected to ddPCR without matched normal counterparts and constitute the TAMARISK validation cohort. Formalin-fixed paraffin-embedded (FFPE) histopathology blocks were obtained, and H&E slides were reviewed by an expert pathologist to score tumor percentage and identify regions of high tumor content as well as regions of normal cells for isolation. Regions were macrodissected from five to ten 10-µm FFPE slides, and DNA was isolated from the excised tissue using the AllPrep DNA/RNA FFPE Isolation Kit (Qiagen, 80234) and the QIAcube according to the manufacturer’s protocols.
Tamoxifen-associated uterine cancer from clinical databases
We identified a TA-UC clinical genomic data cohort by querying cancer registry data at the DFCI. We crossed the diagnosis of UC with the occurrence of breast cancer and tamoxifen treatment, searching for patients who had UC genotype data from the OncoPanel platform70. We identified an overall number of 120 patients, of whom 21 women had primary TA-UC (Extended Data Fig. 6c and Supplementary Tables 1 and 8), diagnosed between 2010 and 2022. A second TA-UC clinical genomic data cohort was obtained using the Caris Life Sciences internal cBioPortal, searching for patients treated with tamoxifen for breast cancer who were later diagnosed with UC. A total of 69 patients were identified, of whom 47 met the criteria for TA-UC, with diagnoses between 2015 and 2023 (Supplementary Table 1 and Extended Data Fig. 6g). Two de novo UC control sets were also identified using the Caris Life Sciences cBioPortal instance: (1) 8,258 patients with primary UC and no prior breast cancer diagnosis and (2) 569 patients with a history of breast cancer but no tamoxifen treatment and primary UC negative for homologous recombination deficiency, identified by the absence of BRCA1 and BRCA2 driver mutations and/or a low genomic scar score71. Genotype data were obtained as previously described72,73. We assessed potential overlap between the two TA-UC clinicogenomic datasets by comparing de-identified clinical variables, including date of UC diagnosis, age at UC diagnosis, histological UC type and prior breast cancer diagnosis. No overlap was found between patients in the two datasets.
Whole-exome sequencing
Whole-exome capture was performed from tumor and normal DNA at the Broad Institute. DNA was quantified in triplicate using a standardized PicoGreen dsDNA Quantitation Reagent (Invitrogen) assay. The quality control identification check was performed using fingerprint genotyping of 95 common SNVs by Fluidigm Genotyping (Fluidigm). Samples were plated at a concentration of 2 ng µl−1 and a volume of 50 µl into matrix tubes, which allowed for positive barcode tracking throughout processing. Samples were sheared using a Broad-developed protocol optimized for a size distribution of ~180 bp. Library construction was performed using the KAPA Library Prep kit with palindromic forked adaptors from Integrated DNA Technologies. Libraries were pooled before hybridization. Hybridization and capture were performed using the relevant components of Illumina’s Rapid Capture Enrichment Kit, with a 37-Mb target. All library construction, hybridization and capture steps were automated on the Agilent Bravo liquid-handling system. After post-capture enrichment, library pools were quantified using qPCR, normalized to 2 nM and denatured using 0.1 M NaOH on the Hamilton STARlet. Flow cell cluster amplification and sequencing were performed according to the manufacturer’s protocols (Illumina) on either the HiSeq 2000 version 3 or HiSeq 2500 runs and used sequencing-by-synthesis kits to produce 76-bp paired reads. The target coverage was 150× mean target coverage for each tumor sample and 60× mean target coverage for each normal sample.
Genomic data alignment and quality control
Data derived from WES were processed using established analytical tools within the Firehose platform (http://www.broadinstitute.org/cancer/cga/Firehose), which was later replaced with a cloud-based platform (FireCloud, Terra) operating on top of the Google Cloud Platform74. These platforms allow for coordinated and reproducible analysis of datasets using analytical pipelines. For each sample, the Picard data processing pipeline (version 2.9.2; http://broadinstitute.github.io/picard/) combines data from multiple libraries and flow cell runs into a single BAM file. Sequencing reads were aligned to the hg19 human genome build using BWA (http://bio-bwa.sourceforge.net). All sample pairs of tumor and normal genotypes were subjected to testing the level of cross-contamination using ContEst version 4 (ref. 75). We calculated the mean sequencing coverage for gene exonic regions using the DepthOfCoverage function from GATK version 4.1.6.0.
Somatic mutation analysis
For each tumor–normal pair, somatic SNVs were called using MuTect (version 1)76 and small insertions and deletions (indels) with Strelka (version 2.9.0)77. These SNVs and indels were annotated using Oncotator (version 1.9.9.0)78. We excluded false-positive SNVs failing the following filters (version 25): (1) the OxoG filter79, which filters sequencing artifacts that are caused by oxidative damage to guanine during shearing in library preparation based on the read pair orientation bias, (2) the FFPE filter80, which filters sequencing artifacts caused by formaldehyde-induced deamination of cytosine based on the read pair orientation bias and (3) a mutational panel of normals81 built from FFPE samples sequenced using the same target regions, allowing us to filter the remaining potential sequencing artifacts as well as germline sites missed in the matched normal tissue. To recover SNVs lost to tumor-in-normal (TiN) contamination from adjacent tissue controls, we applied deTiN (version 3.0)82. In search for the presence of additional mutations (previously observed in TCGA de novo UCs) in the genes ESR1, ESR2, PIK3CA, PIK3R1 and PTEN, we applied a ‘force-calling’ method (version 2)83, which calculates the number of reads supporting an alternate allele at predefined genomic coordinates. Manual review of mutations was performed using the Integrative Genomics Viewer84, and SNVs were filtered due to the following reasons: (1) low allelic fraction (AF) mutations, (2) mutations with orientation bias, (3) mutations called on reads that also contained indels and (4) mutations called in regions with poor mapping. Further downstream analysis was restricted to nonsynonymous mutations, ignoring mutations classified as 3′ UTR, 5′ UTR, IGR, intron, lincRNA, RNA or silent.
Mutational significance analysis
Significance analysis of recurrently mutated genes was performed using MutSig2CV (version 3.11 with ‘gene_min_frac_coverage_required’ set to 0.02), which detects genes with a higher-than-expected SNV frequency or an unexpected pattern of SNVs85. Significantly mutated genes were defined as genes with Q < 0.1 using the method of Benjamini and Hochberg86 to convert final P values to false discovery rate Q values. In addition, we used restricted hypothesis testing (as we have done previously87) using a panel of 113 previously published UC genes (Supplementary Table 4)29,30,31,34 to identify additional recurrently mutated genes. Because our aim was not to perform a de novo discovery of driver genes in the control cohort, we restricted the MutSig2CV analysis in the TCGA sample set of de novo UCs to the above panel of known UC drivers. We tested for mutual exclusivity and co-occurrence on a patient mutational level by applying Fisher’s exact test.
Somatic copy number analysis
GATK4’s copy number variant discovery pipeline was used to analyze read coverage and detect copy number and allelic copy number alterations (release 4.1.6.0; variances of Gaussian kernel for copy ratio segmentation and allele fraction segmentation were set to 0.175 and 0.2, respectively). A copy number panel of normals used normal samples with low TiN to normalize the read depth at each capture probe. In addition, we tagged and removed copy number segments caused by potential germline events by comparing break points and reciprocal overlaps. Manual review of SCNAs was performed using the Integrative Genomics Viewer (version 2.16.2)84.
Copy number significance analysis
GISTIC2.0 (version 2.03.23)36 was applied to detect significantly amplified or deleted SCNAs across a cohort using a threshold of Q < 0.25. Peaks were annotated with genes from the Cancer Gene Census88. G scores were assigned to each peak considering the amplitude of the alteration and the frequency of its occurrence across specimens.
ABSOLUTE, phylogeny and timing analyses
ABSOLUTE version 1.5 (ref. 89) was used to estimate purity (that is, the percentage of tumor cells in the cancer sample), ploidy (that is, the average copy number across the cancer genome), absolute copy numbers and WGD status for each tumor sample. ABSOLUTE solutions were manually curated. To determine whether mutations are clonal (that is, present in all tumor cells), we used the CCF of each mutation provided by ABSOLUTE (mutations with an estimated CCF ≥ 0.95 are considered clonal; mutations with lower CCFs are considered subclonal).
To analyze the phylogenetic relationship between tumor cell populations within a tumor, we used PhylogicNDT (version 35)57,58, an N-dimensional Bayesian clustering framework based on mixtures of Dirichlet processes, in which the number of clusters is inferred over many Markov chain Monte Carlo iterations. Clusters of mutations with consistent CCF were used to determine the phylogenetic tree that best represents the clonal evolution. The tumor developmental trajectory was probabilistically determined, allowing us to order and estimate relative timing of clonal events and WGD (SinglePatientTiming and PhylogicNDT LeagueModel for ordering of events across a sample set).
Prediction of microsatellite instability
MSI was predicted using MSIdetect (version 2) as described before90. In short, MSIdetect assigns a probability for every read from a sequenced sample as coming from a tumor with MSI or an MSS tumor and aggregates it over all reads to generate an MSI score. Because the MSI score varies between sequencing platforms, we used normal samples to set the threshold between MSI and MSS patients.
Mutational signature analysis
SignatureAnalyzer (version 0.0.8)91, a Bayesian nonnegative matrix factorization method, was used to extract mutational signatures from SNVs by considering the 96 single-base substitutions within the trinucleotide sequence context. Signatures were then compared with previously described signatures in COSMIC version 3 (https://cancer.sanger.ac.uk/cosmic/signatures). We also applied supervised Bayesian nonnegative matrix factorization implemented for GPUs92 specifying a set of 13 expected COSMIC version 3 signatures (aging: SBS1, SBS5; MSI: SBS6, SBS14, SBS15, SBS20, SBS21, SBS26, SBS44; POLE: SBS10a, SBS10b, SBS14) to infer their contributions.
Analysis of molecular subtypes
To replicate the molecular subtype analysis from TCGA29, we used the following approach. First, samples were assigned to the POLE subtype if they had POLE exonuclease domain mutations and associated mutational signatures (COSMIC signatures SBS10a, SBS10b and SBS14). Next, samples with MSI (MSI subtype) were classified using MSIdetect and then validated by the presence of mutational signatures associated with it93 (COSMIC signatures SBS6, SBS14, SBS15, SBS20, SBS21, SBS26 and SBS44). The remaining samples were categorized into two groups (CIN and genomically stable) based on their copy number pattern. As described previously94, the CIN subtype is characterized by a high rate of deletions. We calculated the fraction of the genome that was deleted by including copy number events of all lengths with a copy number change larger than a given threshold (R1 = 0.36). Because impure samples have a smaller change in copy number than samples with high purity, the threshold was normalized by the inferred purity. Samples were categorized as CIN when the fraction of the deleted genome was larger than a given threshold (R2 = 0.034). Molecular subtyping was applied to TA-UC and de novo TCGA UC where we did not have previous annotations for molecular subtypes; published molecular subtypes were used for endometrial carcinomas29. Above thresholds were determined by analyzing TCGA Uterine Corpus Endometrial Carcinoma data. ABSOLUTE purity data for TCGA samples were used from Taylor et al.95.
Droplet digital PCR
ddPCR was used to detect hotspot mutations in the PIK3CA and ESR1 genes using FFPE-derived DNA from (1) 19 TA-UCs that had undergone WES and had residual DNA and (2) an independent cohort of 39 TA-UC tumors. TaqMan PCR reaction mixtures were assembled from a 2× ddPCR master mix (Bio-Rad) and custom 40× TaqMan probes or primers made specific for each assay (Thermo Fisher Scientific). Assembled ddPCR reaction mixture (25 μl), which included either 5 μl DNA sample or water as a no-template control, was loaded into wells of a 96-well PCR plate. The heat-sealed PCR plate was subsequently loaded onto the Automated Droplet Generator (Bio-Rad). After droplet generation, the new 96-well PCR plate was heat sealed, placed on a conventional thermal cycler and amplified to the end point. After PCR, the 96-well PCR plate was read on the QX100 Droplet Reader (Bio-Rad). The primers applied in this analysis have been validated and described previously96,97. Analysis of the ddPCR data was performed with QuantaSoft analysis software (Bio-Rad) that accompanied the droplet reader. We calculated the AF (in percent) as AF = (count mutant droplets)(count wild-type droplets + count mutant droplets)−1 × 100 and applied a cutoff of >2% AF to reduce FFPE-associated false positives.
Published human datasets
For comparison of histologic subtypes, research data from 40,587 unique UC tumors diagnosed between 1973 and 2015 were obtained from the SEER9 registries (data released April 2018, based on the November 2017 submission). Tumors were distributed among the nine SEER registries as follows: 17% from San Francisco–Oakland, 13% from Connecticut, 16% from Metropolitan Detroit, 4% from Hawaii, 16% from Iowa, 5% from New Mexico, 16% from Seattle, 6% from Utah and 7% from Metropolitan Atlanta. To match the time frame of our cohorts, only tumors diagnosed between 1983 and 2002 were included. Primary site UCs (ICD-0-2 codes C54.0–C54.3, C54.8–C54.9, C55.9) classified as malignant (ICD-0-3 code 3) were used. To conservatively restrict the dataset to de novo UCs, women with breast cancer history (ICD-0-2 codes C50.0–C50.6, C50.8–C50.9) were excluded, as some may have developed TA-UC following prior tamoxifen treatment. Histologic subtypes were categorized as follows: endometrioid endometrial adenocarcinoma (8050, 8140, 8143, 8210, 8211, 8260, 8261, 8262, 8263, 8380, 8381, 8382, 8383, 8384, 8560, 8570); clear cell (8310) and serous adenocarcinoma (8441, 8460, 8461); mixed (8255, 8323); malignant Mullerian mixed tumors or carcinosarcoma (8950, 8951, 8980, 8981); and sarcoma (8890, 8891, 8896, 8930, 8931, 8935, 8933, 8800, 8801, 8802, 8803, 8804, 8805).
Additionally, we used 554 whole-exome sequenced primary de novo UC samples from TCGA for which data on absolute copy number, SNVs, survival, histological subtype and other clinical variables were available from the MC3 TCGA project81 (Extended Data Fig. 3a). CCFs were identified from the ABSOLUTE-annotated MAF file of the Pan-Cancer TCGA project and Haradhvala et al.93 for 536 of 554 TCGA UC samples. Copy number data were retrieved for a whitelisted set of 544 of 554 tumors. We applied the following criteria to identify de novo TCGA UC samples and exclude prior tamoxifen use: (1) 54 patients were annotated as having no prior tamoxifen use, (2) 482 patients had no prior diagnosis of a malignancy, (3) 16 patients had a prior diagnosis of cancer other than a breast malignancy and (4) two patients were diagnosed with breast cancer, but detailed treatment information excluded prior tamoxifen use. This set of 554 TCGA samples was composed of the following histological types: (1) a sample set containing 371 endometrioid endometrial adenocarcinomas, 96 serous endometrial adenocarcinomas and 19 mixed serous and endometrioid tumors from TCGA Uterine Corpus Endometrial Carcinoma29, (2) 52 uterine carcinosarcomas from TCGA-UCS30 and (3) 16 uterine sarcomas from TCGA-SARC31. For 508 of these patients, height and weight data were available, and BMI was calculated by dividing body weight in kilograms by height in meters squared (kg m−2).
In addition, we searched TCGA annotation files and pathology reports to identify patients with UC and a previous history of tamoxifen use and identified two such patients with TA-UC in the TCGA cohort (TCGA TA-UCs TCGA-BG-A0MS and TCGA-IW-A3M6), who were analyzed separately.
Another set of 130 de novo UC specimens (111 endometrioid endometrial adenocarcinomas, 13 serous endometrial adenocarcinomas, three clear cell carcinomas, three not further defined) with available data on BMI as determined above were used from the Clinical Proteomic Tumor Analysis Consortium94.
We also included 834 primary de novo UC specimens with consistent histology and available mutation data from unique patients from the AACR GENIE Project (version 13.0)32 that originated from the DFCI. Patients with TA-UC (as identified at the DFCI and described above) were excluded. The final set included 527 endometrioid and mixed endometrial adenocarcinomas; 165 serous and clear cell tumors; 93 carcinosarcomas; and 49 leiomyosarcomas.
Although overlap between the US de novo UC cohorts (TCGA, GENIE, CARIS) is highly unlikely due to differences in sample origin, diagnosis data, histology and age at diagnosis, the use of de-identified data means that we cannot completely exclude this possibility, which is a limitation of the study.
In addition, somatic mutation sets from the following noncancerous FFPE tissue types were used: (1) normal endometrial tissue62, (2) endometriosis63,64 and (3) atypical hyperplasia65,66.
Finally, we also included histological subtype data from a set of 161 TAMARISK patients with de novo UC28 diagnosed after breast cancer but without prior use of tamoxifen.
Statistics and reproducibility
Statistical analysis and visualization were performed using R (version 4.1.1) in an RStudio environment and Julia (version 1.7.3) in a Jupyter environment. To determine significance, we used Fisher’s exact test (with Monte Carlo simulation for tables larger than 2 × 2, using 106 iterations), the t-test and the Wilcoxon rank-sum test, all two sided unless otherwise indicated. Multiple-hypothesis testing was performed using the method of Benjamini and Hochberg86, which converted the final P values to false discovery rate Q values; Q < 0.1 was considered significant. The strength of associations between variables was analyzed using Pearson’s correlation. Two-sided stratified Fisher’s exact test was used to control for potential confounding variables when analyzing mutation frequency data across multiple subgroups (or strata), providing a combined P value calculated across the strata, with zero-marginal tables excluded from the calculation98,99. No statistical method was used to predetermine sample size. No data were excluded from the analyses. Randomization and blinding were not applicable, as this study involved retrospective analysis of genomic and clinical data.
Power calculations
We assessed the statistical power to detect differences in driver gene mutation frequencies (either higher or lower) between the TA-UC and de novo UC sample sets given the observed sample sizes in both the WES discovery cohort and the WES validation subtypes. We identified powered genes by computing Bonferroni-corrected two-sided optimal Fisher’s exact test P values across all possible 2 × 2 contingency tables, maintaining the same marginal totals but allowing zero counts. For each configuration, we calculated P values, focusing on the smallest P value as an indication of the extreme case in which the effect size is close to or equal to zero. A Bonferroni-corrected optimal P value of <0.05 was considered a powered test. We also calculated the power to identify driver genes that are significantly less mutated in the TA-UC discovery cohort by computing P values from one-sided Fisher’s exact tests for the different frequencies. Genes at a threshold of P < 0.05 can potentially be considered significantly less mutated in the TA-UC discovery cohort, as they are mutated in at least 76 de novo TCGA UC samples.
Analysis of human expression data
We used previously published100 gene expression levels from Affymetrix U95A Human Genome arrays of enriched human-derived endometrial cells that were short-term cultured with either E2 (100 nM) or tamoxifen (5 µM) for 3 h. After removal of one outlier sample (GSM65291), we performed quantile normalization followed by differential gene expression using limmaVoom101 (version 3.50.0), focusing on genes in the KEGG PATHWAY Database, estrogen response genes from the hallmark gene sets and genes in the AKT–mTOR oncogenic signature gene sets (all from GSEA). Pathway analysis was carried out using Enrichr (https://maayanlab.cloud/Enrichr/)102, the NCI–Nature Pathway Interaction Database103 and differentially expressed genes with a cutoff of |log2 (FC)| > log2 (1.5) and Q value < 0.01.
In vivo mouse study
All mice were maintained in accordance with local guidelines, and therapeutic interventions were approved by the Animal Care and Use Committee of the DFCI (protocol 08-023). To mimic the postmenopausal condition that is typically observed in patients with TA-UC, 20 C57BL/6 female mice (Jackson Laboratory) were oophorectomized after sexual maturity (6–7 weeks) to allow for proper uterine development. Oophorectomy also circumvented the ER-dependent endometrial changes that occur during the estrous cycle, which could confound the interpretation of results. As the hormone E2, a major female sex hormone produced during the estrous cycle, binds to ER and increases cell proliferation, we used exogenous E2 as a positive control. Mice were randomized (n = 5 per arm) to E2 (0.01 mg per pellet, 60-d release), vehicle control (E2 deprived), tamoxifen (Sigma, in 20% ethanol in corn oil, 0.5 mg per mouse per day, subcutaneous injection, comparable to the concentration seen in humans104) or tamoxifen plus alpelisib (Selleckchem, in 30% PEG 400 + 0.5% Tween-80 + 5% propylene glycol, 30 mg per kg per day, oral gavage) for 30 d. At the end of the study, mice were euthanized, and uterine horns were collected.
Mouse tissue collection and processing
Mouse uterine horns were collected from five mice per cohort, as reported by De Clercq et al.105. Samples were allocated for downstream applications as follows: (1) single-cell suspensions were prepared and used to isolate epithelial and stromal cell populations. For the E2, tamoxifen and tamoxifen-plus-alpelisib groups, three mice per condition were used; in the vehicle control group, five mice were processed to obtain sufficient material despite the minuscule size of the uteri in this condition. (2) FFPE samples for IHC were prepared from three mice (E2), five mice (tamoxifen, tamoxifen plus alpelisib) and two mice (vehicle control, in which sample collection was limited by the miniscule size of the uterine horns, a consequence of oophorectomy without hormonal supplementation, and by fibrosis secondary to the surgical procedure).
Immunohistochemistry
For immunohistochemical detection, samples were stained with primary antibodies and incubated with anti-mouse (G21040, Invitrogen) or anti-rabbit (G21234, Invitrogen) antibodies (both at a 1:2,000 dilution) for 50 min at room temperature. Samples were stained with the DAB (3,3′-diaminobenzidine) colorimetric substrate and counterstained with hematoxylin. The following primary antibodies were used: anti-ER-α (06-938, 1:1,000, Millipore), anti-phospho-IR/IGF1R Tyr1162/Tyr1163 (44-804, 1:500, Invitrogen), anti-Ki-67 (ab15580, 1:1,000, Abcam), anti-phospho-AKT Thr308 (ab81283, 1:50, Abcam) and anti-phospho-S6 Ser240/Ser244 (2215, 1:500, Cell Signaling).
Numbers of ducts per mouse were counted in six distinct sections using a 20× high-power field. The length (in µm) of endometrial epithelial cells per mouse was measured in six sections using five distinct regions of the internal lumen. IHC images were analyzed with QuPath version 0.2.0 software (https://qupath.github.io/). IHC staining was quantified as the product of percent positive cells per section × staining intensity in optical density (H score). Statistical analyses for immunohistochemical studies were performed in GraphPad Prism version 9.0 (GraphPad Software) using one-way ANOVA.
Messenger RNA in situ hybridization
In situ hybridization was performed with the RNAscope Intro Pack for Multiplex Fluorescent Reagent Kit v2-Mm from Advanced Cell Diagnostics according to the manufacturer’s protocol. Briefly, FFPE sections were deparaffinized with xylene and rehydrated with alcohol. The sections were hybridized at 40 °C for 2 h with the RNAscope Probe-Mm-Igf1 that is specific for mouse Igf1 mRNA (Advanced Cell Diagnostics), and the signal was visualized with RNAscope fluorescent reagents. Sections were counterstained with ProLong Gold Antifade Reagent (Life Technologies) before dehydrating, and coverslips were affixed with Permount (Thermo Fisher Scientific). Images were acquired with a Leica SP8X STED/confocal microscope using Leica Application Suite X (version 3.7) acquisition software. Images were acquired as Z stacks (1 µm) using the Piezo Z stage.
RNA extraction and quantitative PCR with reverse transcription
Total RNA was isolated using TRIzol (Life Technologies) and the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. To test the purity of epithelial cells, we used quantitative PCR with reverse transcription and primers summarized in Supplementary Table 15. mRNA was retrotranscribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystem), and detection was accomplished using the Roche LightCycler 480 Real-time PCR system in combination with the Power SYBR Green PCR Master Mix (Life Technologies).
RNA sequencing
RNA-seq libraries were made after enrichment with oligo(dT) beads. First, mRNA was randomly fragmented by adding fragmentation buffer. Next, cDNA was synthesized using mRNA template and random hexamer primers, after which a custom second-strand synthesis buffer (Illumina), dNTPs, RNase H and DNA polymerase I were added to initiate second-strand synthesis. After a series of terminal repair, A ligation and sequencing adaptor ligation, the double-stranded cDNA library was completed through size selection and PCR enrichment. Samples were sequenced on an Illumina NextSeq 500 instrument (libraries generated and sequencing performed at Novogene).
RNA sequencing analysis
RNA-seq analysis was performed using the VIPER analysis pipeline (version 1.41.0)106. Alignment to the hg19 human genome was accomplished using STAR version 2.7.0f followed by transcript assembly using cufflinks version 2.2.1 (ref. 107) and RSeQC version 2.6.2 (ref. 108). Differential expression analysis was carried out using DESeq2 version 1.18.1 (ref. 109). Pathway analysis was carried out using Enrichr (https://maayanlab.cloud/Enrichr/) and applying MsigDB oncogenic signatures102.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.