The hallmark of HIV infection is progressive CD4+ T lymphocyte depletion, resulting in persistent immunodeficiency that markedly increases susceptibility to opportunistic infections, particularly pulmonary complications [8]. Our findings demonstrate significantly reduced CD4+ T cell count in HIV-infected patients compared to controls (P < 0.05), confirming this immunopathological basis. Notably, even with effective ART, HIV infection sustains chronic immune activation and inflammatory responses involving T cells, B cells, and monocytes, accompanied by elevated pro-inflammatory cytokines including TNF-α, IL-6, and acute-phase proteins such as CRP [9,10,11,12,13,14]. This persistent inflammatory state not only contributes to non-AIDS-related morbidity and mortality [13, 14], but may also fundamentally alter inflammatory responses to acute secondary infections, such as the pulmonary infections central to this investigation.
As a key acute-phase protein, SAA rapidly escalates following inflammatory stimuli and orchestrates pathogen clearance by recruiting monocytes, neutrophils, dendritic cells, and T cells to sites of inflammation [15]. In chronic inflammatory states, circulating SAA levels—though lower than acute-phase peaks—remain elevated above baseline [16]. However, there is limited research on the dynamic changes of SAA in HIV-infected individuals, particularly in cases of concurrent acute infections (e.g.pulmonary infections), and its diagnostic value remains underexplored [17]. This study therefore addresses a critical gap by examining how chronic HIV infection modulates SAA responsiveness to acute pulmonary inflammation.
A pivotal observation was the markedly higher acute-phase SAA and CRP levels in HIV-positive patients with lung infections versus non-HIV controls (P < 0.05), aligning with Hussbekk et al. [18]. We propose two mechanistic explanations: (1) The compounding effects of chronic immune activation: HIV-induced low-grade inflammation may hypersensitize the immune system to secondary lung infections, amplifying acute-phase protein release; (2) Increased pathogen burden: Immunodeficiency likely predisposes HIV patients to heavier or polymicrobial infections, intensifying the acute-phase response. Moreover, this study demonstrated significantly elevated serum LDH levels in HIV-infected patients compared to controls (P < 0.05), indicating exacerbated tissue damage during concurrent pulmonary infection. Given the well-documented sensitivity of LDH in HIV-associated Pneumocystis pneumonia (PJP) [19], these findings further corroborate that pulmonary infection intensifies histopathological injury in HIV-positive individuals. Together, the data explain the prolonged hospitalization observed in these patients (P < 0.05).
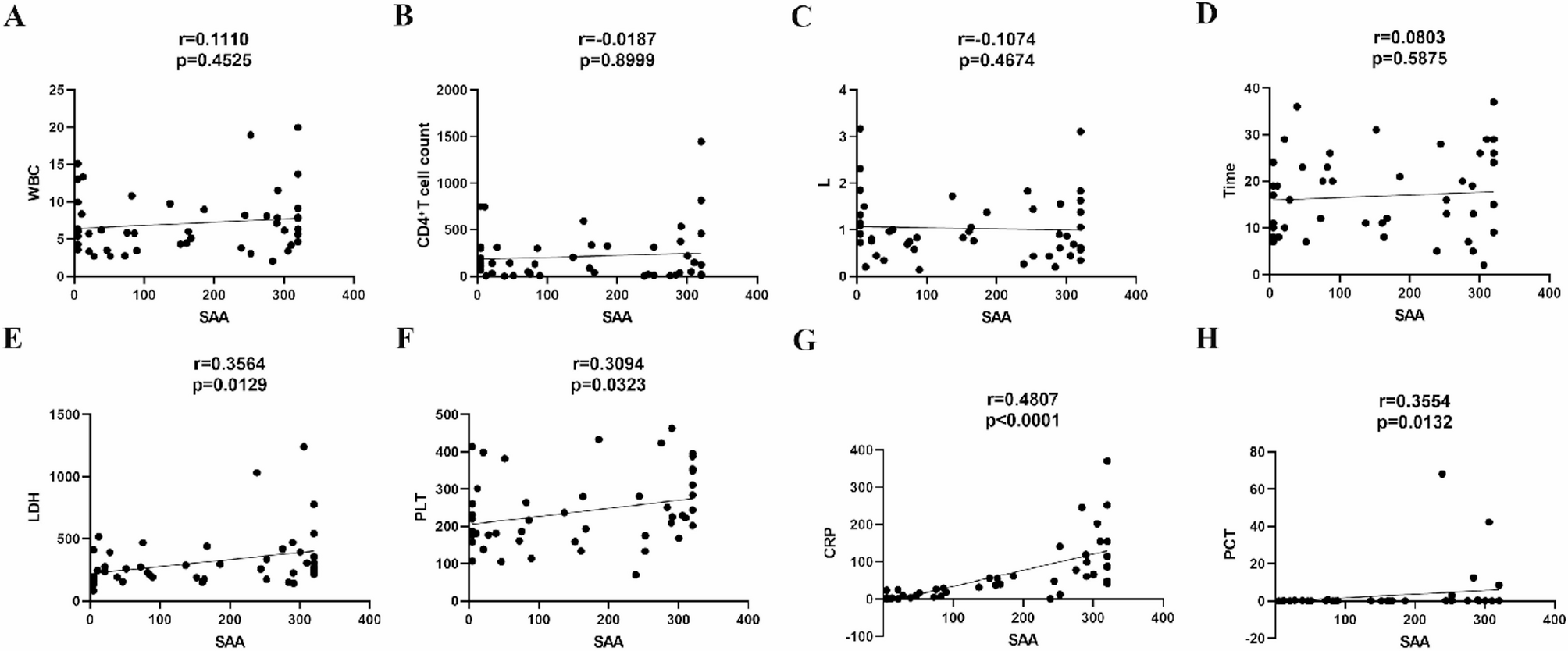
In analyzing inflammatory markers, we observed that SAA levels in HIV patients with pulmonary infections showed significant positive correlations with CRP, PCT, LDH, and PLT. This finding holds important clinical implications. As CRP and SAA are among the most sensitive acute-phase proteins, their concentrations are strongly associated during inflammation, with SAA typically exhibiting a more pronounced increase [20,21,22]. PCT, driven by endotoxin and cytokines, signals bacterial involvement. LDH serves as a highly sensitive biomarker for tissue injury, reflecting cellular damage with remarkable precision. Beyond its well-known role in hemostasis, emerging evidence highlights PLT as pivotal “first responders” in infection immunity. They actively combat pathogens through direct microbicidal action and by orchestrating immune cell-mediated pathogen clearance [23, 24]. These linkages position SAA as a integrative biomarker reflecting inflammatory intensity, tissue injury, and immune engagement in HIV-lung coinfection. In clinical practice, this approach offers a more effective predictive tool for early diagnosis of acute infections in patients with prolonged chronic infections. Furthermore, the study revealed no significant correlation between SAA levels in HIV patients with pulmonary infections and either CD4+ T cell counts or hospitalization duration. This may be attributed to SAA’s nature as a highly sensitive but short-lived inflammatory marker—it surges rapidly within 24 h of stimulation and declines swiftly post-acute phase. Consequently, while it detects inflammation effectively, it shows minimal association with long-term clinical outcomes such as hospital stay [25, 26].
Beyond acute-phase roles, SAA participates in tissue remodeling, atherosclerosis, and cancer metastasis via receptor interactions and metalloproteinase modulation [1, 27], suggesting potential implications for HIV-related chronic complications—a avenue warranting further study.
This study has several potential limitations. First, as a retrospective analysis, it inherently carries a lower level of evidence compared to prospective studies. Second, although all enrolled patients presented with pulmonary infections, the causative pathogens varied substantially, leading to significant microbial heterogeneity. Third, incomplete viral nucleic acid data in the HIV group precluded further analysis of the relationship between infection and viral load. Fourth, the modest sample size (approximately 50 patients) may limit the robustness of the key findings. Despite these limitations, this study represents the first investigation into the role of SAA in HIV patients with acute lung infections, providing a foundation for future research. Addressing these shortcomings will be a priority in subsequent, more in-depth studies.