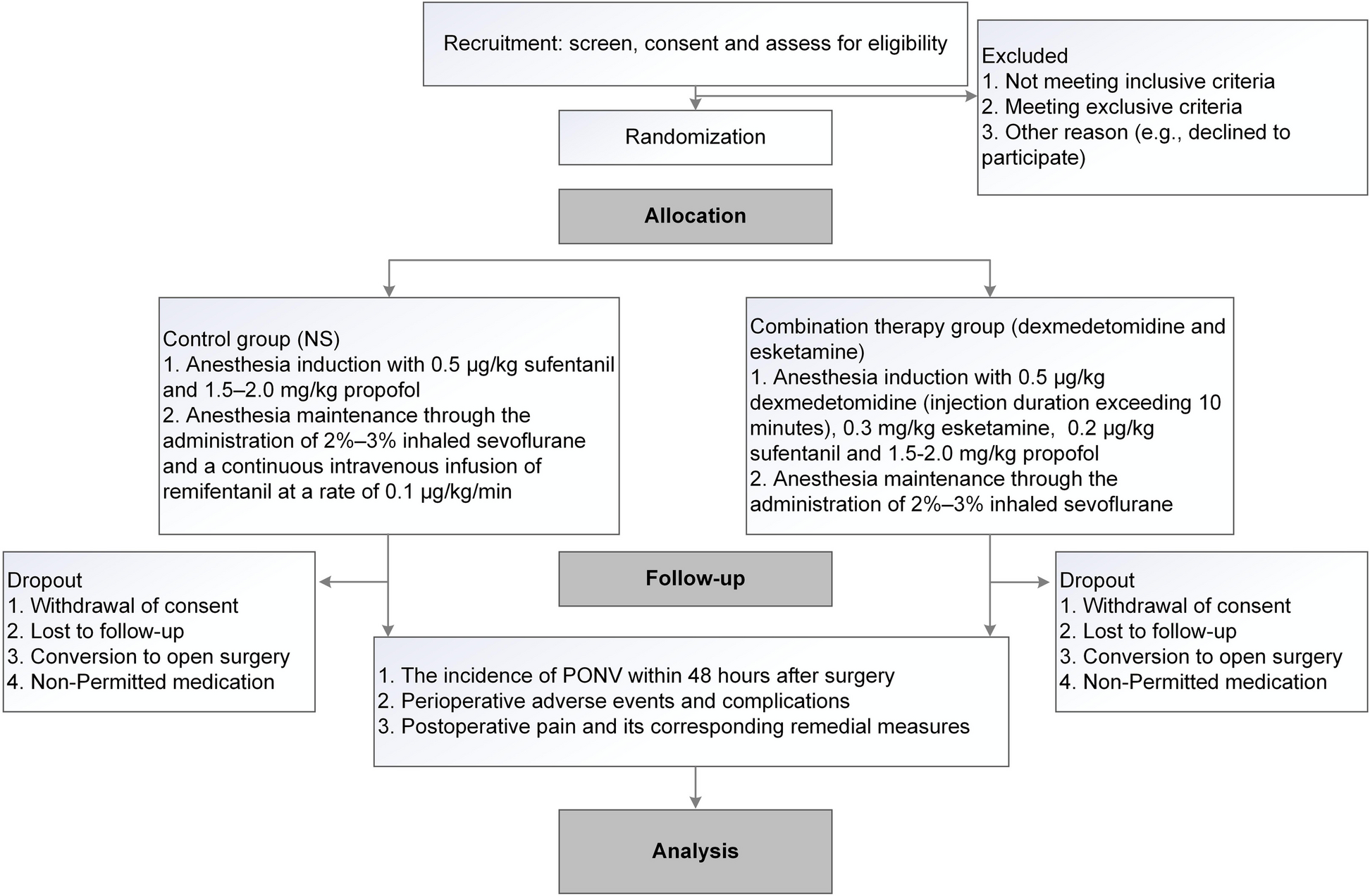
Study setting {9}
The investigation will be carried out by the department of anesthesiology at Suzhou Ninth People’s Hospital, an affiliated hospital of Soochow University. Our hospital’s anesthesiology department is recognized as a key clinical discipline in the region. With around 5000 laparoscopic surgery patients treated annually, the hospital will provide a sufficient patient population to ensure an adequate sample size for the study. Figure 1 illustrates the research workflow.
Study flow diagram. PONV, postoperative nausea and vomiting
Eligibility criteria {10}
Inclusion criteria
-
1.
Participants aged 18 to 65 years old.
-
2.
American Society of Anesthesiologists (ASA) physical status I to III.
-
3.
Body mass index (BMI) between 18 and 30 kg/m2.
-
4.
Scheduled to perform general anesthesia with endotracheal intubation for laparoscopic surgery, including appendectomy, laparoscopic cholecystectomy and laparoscopic hernia repair.
-
5.
Expected duration of surgery between 30 and 120 min.
Exclusion criteria
-
1.
Sick sinus syndrome or severe bradycardia (heart rate less than 50 beats per minute).
-
2.
History of hypertension or cardiac insufficiency.
-
3.
Second-degree or higher atrial block without a pacemaker.
-
4.
Left ventricular ejection fraction less than 40%.
-
5.
Diagnosed with coronary artery disease or history of myocardial infarction.
-
6.
Hepatic or renal insufficiency, Child–Pugh class C, or undergoing renal replacement therapy.
-
7.
Parkinson’s disease or Alzheimer’s disease.
-
8.
Seizures or epilepsy.
-
9.
Current pregnancy or lactation status.
-
10.
History of persistent pain or prior use of sedatives or analgesics.
-
11.
Known allergies to the drugs used in this study.
-
12.
Participation in another clinical trial within the past 30 days.
-
13.
History of substance abuse.
-
14.
Psychiatric illness or current use of antipsychotic medications.
-
15.
Communication disorders such as deafness or cognitive impairment.
-
16.
Anticipated difficult airway or history of difficult intubation.
Drop out criteria
Participants will not be excluded from the final analysis solely due to adverse events or other post-randomization occurrences. All randomized participants will be included in the intention-to-treat (ITT) analysis.
However, the following circumstances will be considered as dropouts, and the reasons will be recorded in detail:
-
1.
Withdrawal of informed consent for continued participation or data use.
-
2.
Loss to follow-up before the assessment of primary or secondary outcomes.
-
3.
Conversion from laparoscopic to open surgery.
-
4.
Use of non-permitted medications, including:
-
– Additional antiemetics not specified in the protocol.
-
– Perioperative corticosteroids.
-
– Sedatives or analgesics outside the study regimen.
-
-
5.
Non-collection of data.
In contrast, the following events will be considered protocol deviations and addressed in sensitivity analyses:
-
1.
Non-administration of the study drug.
-
2.
Unplanned additional surgical procedures.
-
3.
Minor violations of timing or dosage not affecting outcome measurement.
Screening failures (participants who do not meet eligibility criteria before randomization) will be recorded separately and excluded from all analyses. All dropout events and reasons will be meticulously documented in the case report forms (CRFs) and stored for auditing and future reference.
Consent or assent {26a, 26b}
Eligible patients will be approached by research team members, all of whom are licensed medical doctors, to be invited to participate in the study. Detailed instructions regarding the study protocol, procedures, and potential risks and benefits will be provided in clear language. Written informed consent will be obtained from each participant one day prior to surgery to ensure voluntary participation and adequate understanding of the research process.
Explanation for the choice of comparators {6b}
To provide a rigorous comparison, the comparator in this trial is the standard anesthetic regimen routinely used at our institution, consisting of intravenous induction with sufentanil and propofol, followed by maintenance with sevoflurane and a continuous remifentanil infusion. This approach is widely adopted in clinical practice and provides effective intraoperative analgesia with minimal postoperative sedation, making it particularly suitable for laparoscopic surgeries [11, 29]. The intervention group protocol is informed by the principles of OFA, where dexmedetomidine and esketamine are commonly utilized to achieve adequate analgesia and sedation while minimizing opioid exposure. The selected drug combination and administration strategy are based on published OFA studies and adapted for routine clinical application [13, 21, 25]. Given the established link between intraoperative opioid use and PONV, this study aims to determine whether an opioid-reducing approach incorporating these agents can improve PONV outcomes and postoperative recovery compared to the standard opioid-based regimen.
Interventions {11a, 11b, 11c, 11d}
Patients will be randomized into two groups using a computer-generated random number table at a 1:1 ratio, comprising the combination therapy group (dexmedetomidine and esketamine) and the control group. In the combination therapy group, anesthesia induction will involve intravenous infusion of dexmedetomidine (0.5 μg/kg over more than 10 min), followed by intravenous bolus administration of esketamine (0.3 mg/kg), sufentanil (0.2 μg/kg), and propofol (1.5–2.0 mg/kg). Anesthesia will be maintained with 2–3% sevoflurane. In the control group, anesthesia induction will consist of intravenous bolus administration of sufentanil (0.5 μg/kg) and propofol (1.5–2.0 mg/kg), while maintenance will include 2–3% sevoflurane inhalation and continuous intravenous infusion of remifentanil at 0.1 μg/kg/min. The selected remifentanil dose is within the low range and below thresholds typically associated with remifentanil-induced hyperalgesia, as supported by previous studies [30, 31].
All patients will receive 5 mg of dexamethasone intravenously after anesthesia induction, 4 mg of tropisetron at the end of surgery, and 50 mg of flurbiprofen axetil approximately 30 min before surgery completion to prevent PONV and manage postoperative pain. The intraoperative monitoring protocol includes ECG, SpO2, non-invasive blood pressure, and end-tidal CO₂, with anesthesia depth maintained within a BIS range of 40–60. Vital signs will be continuously monitored using standard multi-parameter monitors. The dosages of anesthetic agents in both groups are derived from published literature and institutional protocols, with remifentanil and sufentanil dosing in the control group based on perioperative anesthesia studies [13, 32], and dexmedetomidine and esketamine dosing in the combination group adapted from opioid-free anesthesia protocols [13, 21].
Postoperative management will also be standardized. Pain and PONV assessments will be conducted at fixed time intervals: 0–6 h (PACU), 6–24 h, and 24–48 h after surgery. Pain intensity will be evaluated using the numerical rating scale (NRS) at 0, 6, 12, 24, and 48 h. Time to first PONV episode, time to first rescue medication use, and total dosage/frequency of rescue analgesics and antiemetics within 48 h will be recorded. Postoperative adverse events such as nightmares, drowsiness, bradycardia, length of hospital stay, and discharge condition will also be documented.
Rescue interventions are standardized. For pain (NRS ≥ 4), 5 mg of dezocine will be administered intravenously. Severe PONV, defined as ≥ 3 vomiting episodes or inability to perform daily activities, will be managed with 10 mg of azasetron; persistent vomiting post-treatment may lead to study withdrawal. Adverse reactions such as esketamine-related nightmares will be treated with 2 mg midazolam, while drowsiness will be managed with observation or opioid antagonists like naloxone in severe cases. Bradycardia (HR < 55 bpm) will be treated with 0.25 mg atropine or 2 μg isoproterenol and anesthetic dose adjustment.
To enhance adherence and consistency, interventions will be performed under general anesthesia, eliminating the need for patient cooperation during drug administration. Postoperative assessments will be carried out by trained personnel at predefined time points using standardized tools. Rescue medication criteria are clearly defined to minimize variability. Preoperative briefings for nursing and anesthesiology teams will ensure uniform postoperative care. The overall trial process, including patient enrollment, treatment, and data collection, will follow the SPIRIT guidelines as detailed in Table 2.
Outcomes {12}
Primary outcome
Postoperative pain and nausea severity will be assessed using the Numerical Rating Scale (NRS), an 11-point scale ranging from 0 (no symptom) to 10 (worst imaginable pain or nausea), based on patient self-report at predefined postoperative intervals. The incidence of nausea will be defined as any self-reported score ≥ 1, while vomiting will be defined as any observed or self-reported episode of forceful expulsion of gastric contents. Both nausea and vomiting episodes will be recorded separately by trained clinical staff through direct observation and/or patient reports. The primary outcome of this study is the incidence of PONV (including both nausea and vomiting) within 48 h after surgery. PONV will be assessed by trained clinical staff during three defined time intervals: 0–6 h, 6–24 h, and 24–48 h postoperatively. Both nausea and vomiting episodes will be recorded separately to allow for detailed analysis.
Secondary outcomes
Preoperatively, the Apfel simplified risk score will be used to evaluate each patient’s baseline risk of PONV, assigning one point for each of the following: female sex, non-smoking status, history of motion sickness or previous PONV, and anticipated postoperative opioid use (total score range: 0–4). The secondary outcomes include:
-
1.
Preoperative Apfel PONV risk score.
-
2.
NRS pain scores of the patients recorded at 0 h (in the PACU), 6 h, 12 h, 24 h, and 48 h after surgery.
-
3.
Time to first PONV episode and time to first rescue antiemetic administration.
-
4.
Time to first rescue analgesic administration.
-
5.
Total dosage and frequency of rescue analgesics and antiemetics within 48 h.
-
6.
Patient satisfaction score at discharge, rated on a 5-point Likert scale.
-
7.
Length of hospital stay (in days).
-
8.
Discharge condition score, assessed by the attending physician.
-
9.
Incidence and classification of AEs.
Participant timeline {13}
The timeline for participant involvement is illustrated in Table 2.
Sample size calculation {14}
In a recent investigation, OFA demonstrated a 65% reduction in the likelihood of PONV following laparoscopic gynecological surgery, decreasing the incidence from 42.5 to 15.0% (10). For the power analysis, we assume a baseline PONV incidence of 40% in laparoscopic surgery under traditional opioid anesthesia. With the hypothesis of a 50% average reduction in PONV, our combination therapy strategy is anticipated to lower the PONV incidence to 25%. To achieve a statistical power of 80% with a bilateral α level of 0.05, 64 patients per group will be deemed necessary to detect intergroup differences in PONV. Considering potential withdrawals, a planned recruitment of 140 patients will be intended, with 70 in each group. The sample size is determined using PASS software (V.11.0.7, NCSS, Kaysville, UT, USA).
Recruitment {15}
Patients participating in this study were enlisted from the anesthesia department. Our recruitment information was disseminated through the WeChat public platform. Additionally, soliciting recommendations from medical personnel constituted a significant aspect of our recruitment efforts. Furthermore, recruitment posters were prominently displayed in areas like the hospital outpatient and inpatient departments.
Allocation {16a, 16b, 16c}
In this study, randomization tables will be generated using IBM SPSS Statistics version 26.0 and maintained by independent statisticians overseeing the trial. Eligible participants will be randomly allocated to either the combination therapy group or the control group in a 1:1 ratio. To ensure allocation concealment, the randomization assignments will be enclosed in sealed, opaque envelopes and securely stored in a designated office. Neither the participants nor the investigators involved in clinical care or outcome assessment will be informed of group assignments, thereby maintaining the double-blind design. In cases of emergency or where clarification is required, only designated researchers will have access to the allocation list. Before surgery, coded study medications will be distributed to study personnel. The allocation sequence will remain confidential, accessible only to the principal investigator (PI) and an independent, unblinded researcher responsible for study drug preparation. The PI will assign participants to treatment groups according to the randomization list, while the unblinded researcher, who will not be involved in drug administration, anesthesia management, or outcome assessment, will prepare the corresponding study medications.
Blinding {17a, 17b}
To maintain double-blinding, each study syringe will be labeled solely with the participant’s unique identification number, without revealing the group allocation. The unblinded researcher, who is not involved in clinical care or outcome assessment, will prepare the study medication according to the randomization list and ensure that all other clinical staff and participants remain blinded throughout the trial. To ensure identical appearance and preserve blinding, both dexmedetomidine and esketamine (or their corresponding placebo components) will be diluted with 0.9% normal saline to a total volume of 20 ml. The final preparations, which are colorless and transparent, will be loaded into identical 20 ml syringes and handed over to the anesthesiologist immediately prior to anesthesia induction. All study personnel involved in clinical care, anesthesia management, outcome assessment, and data collection, as well as the participants themselves, will remain blinded to treatment allocation until data collection is completed and final analyses are conducted. In the event that unblinding is necessary due to medical emergencies or other justified reasons, access to allocation information will be strictly limited to designated personnel responsible for medication distribution.
Data collection methods {18a, 18b}
The subsequent data will be gathered as follows:
Preoperatively
-
1.
Patient’s general information (including height, weight, ASA classification, level of education, smoking habits, history of motion sickness, previous opioid use, allergies and surgical history).
-
2.
Apfel PONV risk score.
-
3.
Baseline NRS pain score.
-
4.
Results of laboratory tests.
Intraoperatively
-
1.
Hemodynamic parameters (such as NBP, HR, ECG and SpO2).
-
2.
Surgical details (including duration of operation, anesthesia, pneumoperitoneum, dosage and concentration of anesthetic agents administered, blood loss, volume of fluid replacement, urine output, and body temperature).
Post-surgery
-
1.
Incidence of nausea and vomiting will be assessed at three intervals: 0–6 h (in the PACU), 6–24 h, and 24–48 h after surgery.
-
2.
NRS pain scores will be recorded at 0 h (PACU), 6 h, 12 h, 24 h, and 48 h postoperatively.
-
3.
Time to first PONV episode, time to first rescue antiemetic or analgesic administration, and the total dosage and frequency of rescue medications.
-
4.
Incidence of AEs.
-
5.
Postoperative laboratory results.
-
6.
Length of hospital stay and discharge condition.
All patient data will be meticulously recorded in a case report form by a designated independent researcher. These records will then be entered into an electronic database under the careful supervision of the PI. Oversight of data collection will be managed by a Data Monitoring Committee (DMC), with final analysis conducted by impartial statisticians.
To promote participant retention and ensure complete follow-up, investigators will provide a comprehensive explanation of the study protocol and expected outcomes during the preoperative assessment. Efforts will be made to maximize participants’ understanding of the study procedures through detailed instruction and clear guidance, thereby enhancing their compliance and engagement throughout the study period.
Data management {19}
Prior to commencing the study, members of our trial team will undergo training in the collection, management, storage and confidentiality of data to ensure comprehension and compliance with pertinent policies and regulations. Patient data will be securely stored in both paper and electronic formats. Coded paper records will be kept in designated, locked storage areas. Data entry for the study will be conducted using a password-protected Microsoft Access database by two trained team researchers employing a double-entry method, and the accuracy of entries will be verified against the electronic database. To minimize the risk of data loss, researchers will perform incremental backups on a daily basis.
Statistical analysis {20a, 20b, 20c}
The Shapiro–Wilk test will be employed to assess the normality of data distribution. Data will be presented as mean (standard deviation), median (interquartile range), or number (percentage), as appropriate. Descriptive statistics will be used primarily to summarize patient characteristics and baseline variables. Comparative analyses of perioperative variables and outcome measures will be performed using the Mann–Whitney rank sum test, chi-square test, or Fisher’s exact test, depending on data type and distribution. To evaluate the effect of combination therapy versus control, the median difference (MD) or odds ratio (OR) with corresponding 95% confidence intervals (CI) will be calculated.
Subgroup analyses of the primary outcome (PONV incidence) will be conducted based on gender, smoking status, and Apfel PONV risk score. No adjustments will be made for multiple testing in secondary outcome analyses, which will therefore be interpreted as exploratory. All statistical analyses will be conducted using IBM SPSS software (version 19.0; IBM SPSS, Chicago, IL, USA), with two-sided P-values < 0.05 considered statistically significant.
As the administration of study medications will be supervised by anesthesiologists, protocol adherence is expected to be high. Since outcome assessment is scheduled within 48 h postoperatively, the occurrence of missing primary outcome data is anticipated to be minimal. Any missing data will not be imputed.
Data monitoring {21a, 21b}
The data monitoring process for this study will be overseen by the monitoring manager, who is a member of the clinical trial management team at Suzhou Ninth Hospital Affiliated to Soochow University. This individual will be responsible for ensuring the proper preservation of informed consent documents, monitoring participant compliance, and verifying the validity and safety of the study data throughout the trial. Given the short duration of the study, the relatively small sample size, and the anticipated low incidence of serious adverse events (SAEs), no interim analysis is planned.
Harms {22}
All adverse events (AEs) will be closely monitored and documented throughout the perioperative period and until the patient is discharged from the hospital. Based on previous studies [27, 28], these include the following:
-
1.
Cardiovascular events: hypotension (systolic blood pressure < 90 mmHg), hypertension (systolic blood pressure > 180 mmHg), bradycardia (heart rate < 50 bpm), tachycardia (heart rate > 120 bpm), arrhythmias.
-
2.
Respiratory events: respiratory depression (respiratory rate < 8 breaths/min or SpO2 < 90%), apnea, bronchospasm.
-
3.
Neurological and psychiatric events: emergence delirium or agitation, dizziness, headache, visual or auditory hallucinations, excessive sedation (BIS < 40 or unresponsiveness), seizures.
-
4.
Injection site reactions or hypersensitivity: rash, pruritus, swelling, or anaphylaxis.
Each AE will be classified by severity into mild, moderate, or severe according to the Common Terminology Criteria for Adverse Events (CTCAE) v5.0.
Severe adverse events (SAEs) are defined as unanticipated medical incidents that prolong hospitalization, result in persistent disability or dysfunction, pose a life-threatening risk, or cause death. If any SAE occurs, the infusion of dexmedetomidine and esketamine will be immediately discontinued, and the participant will be withdrawn from the study if necessary. All SAEs will be reported promptly to the Ethics Committee, and participants will be followed until the event resolves or stabilizes, or until hospital discharge, whichever comes later.
The attending anesthesiologists and trained research staff will be responsible for managing and recording all AEs and SAEs in the case report forms. An independent Data Monitoring Committee (DMC), composed of clinical experts not involved in the study, will review and categorize all AEs and SAEs according to predefined criteria. If a participant experiences more than three episodes of vomiting despite rescue treatment, or develops any SAE, the case will be considered for withdrawal from the study in accordance with the predefined discontinuation criteria.
Auditing {23}
There will be no plans for conducting formal trial audits.
Research ethics approval {24}
Ethical clearance for this investigation was granted by our hospital’s ethics committee on May 1, 2023 (2,023,067). Subsequently, the research protocol was registered with the China Clinical Trial Registry on June 14, 2023 (ChiCTR2300072455).
Protocol amendments {25}
Should there arise a need for protocol modifications, they will be duly registered at https://www.chictr.org.cn.
Confidentiality {27}
Confidentiality will be maintained for all potential and enrolled patients, with access restricted solely to the principal investigator. Anonymized patients will be assigned unique numerical identifiers (ID numbers) rather than names. Throughout the duration of the experiment, the DMC diligently oversees the database to enhance data integrity. Upon completion of the experiment, researchers will procure the results of statistical data analysis.