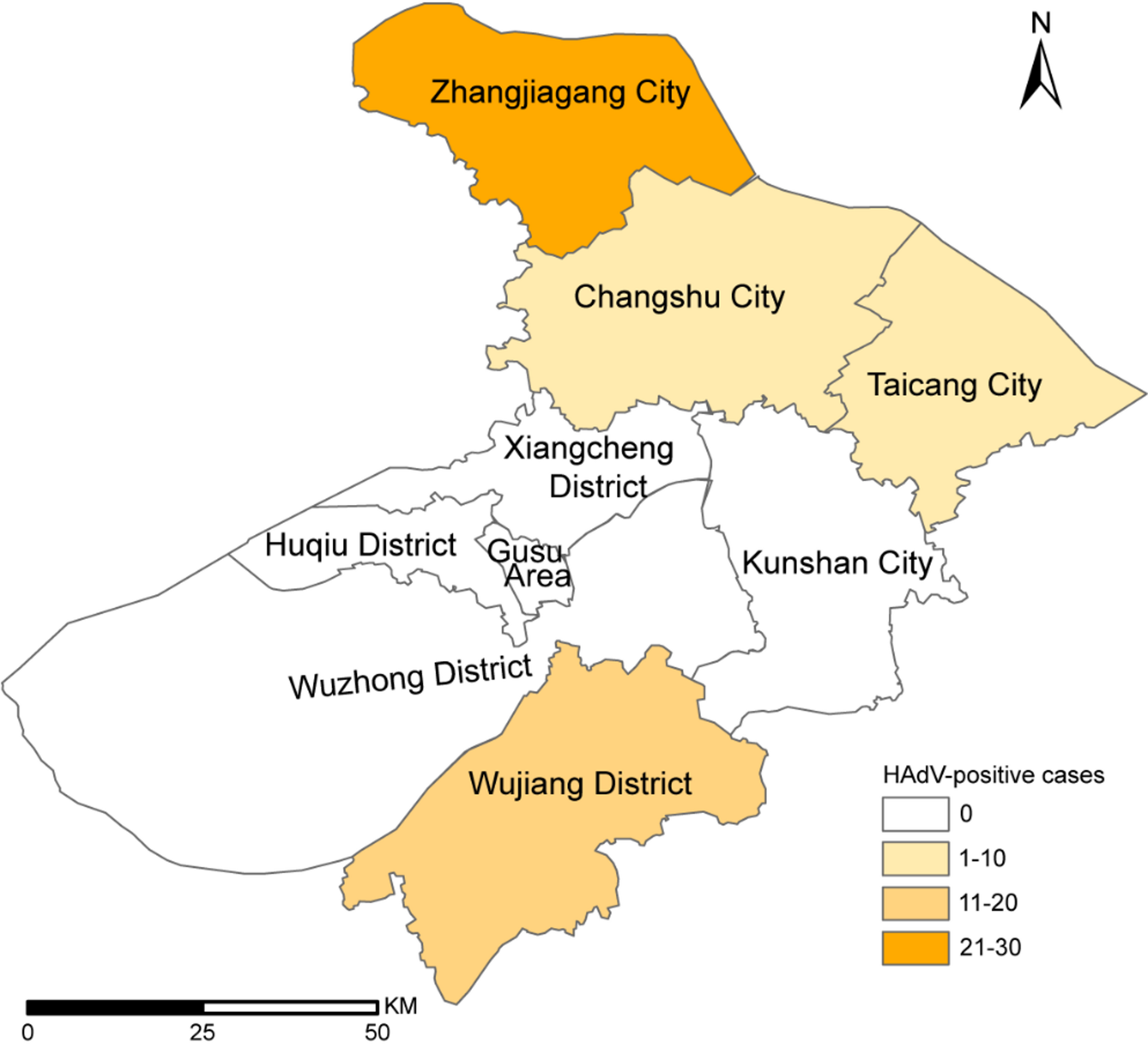
Case information
Among the 488 throat swab samples collected from settings with ILI outbreaks in Suzhou between March 2024 and January 2025, 53 tested positive for HAdV via real-time PCR. These cases were reported in May (25 cases), June (10 cases), November (8 cases) and December (10 cases). The overall HAdV positive rate was 10.86%, ranking second among common respiratory pathogens, following influenza virus (36.68%) and preceding SARS-CoV-2 (9.43%), rhinovirus (5.74%) and enterovirus (4.51%). No significant regional difference in HAdV-positive rates was observed across Zhangjiagang City (22.43%), Wujiang District (33.33%), Taicang City (40%) and Changshu City (20%) (Fig. 1 and Table 1). However, there was a slight difference in gender distribution, with a higher HAdV-positive rate in males than in females (13.83% vs. 7.66%, p = 0.028) (Table 1).
All HAdV-positive cases were exclusively identified in children aged 7–10 years, whereas no infections were detected in the 3–6 year or 11–15 year age groups. To further access the age distribution of HAdV infections, we analyzed data from ILI outpatients and hospitalized pneumonia patients from the same period in Suzhou. As shown in Supplementary Table 1, there were significant differences in HAdV-positive rates across age groups in both cohorts (p < 0.001). Among ILI outpatients, the positive rate peaked in the 7–10 (16.99%) and 3–6 (16.89%) year age groups. Among hospitalized pneumonia patients, the highest positive rate was observed in the 3–6 year age group (10.00%) (unpublished data).
The geographical distribution of HAdV outbreak samples in this study
Phylogenetic analysis
In this study, 29 samples were selected from the 53 HAdV-positive outbreak samples for virus isolation and whole-genome sequencing. Detailed sample information is provided in Supplementary Table 2. The nucleotide identities of the whole genome, hexon, penton base and fiber genes were 97.5–100%, 94.2–100%, 99.5–100% and 97.2–100%, respectively. The phylogenetic tree constructed from whole genome sequences was divided into four clusters based on topological structure and evolutionary distance. The intra-cluster distances ranged from 0 to 0.0371, while the inter-cluster distances ranged from 0.050 to 0.192. All 29 outbreak strains were assigned to Cluster 1, together with all HAdVB3 and HAdVB7 reference strains circulating in the United States, Germany and China between 1955 and 2023. Additionally, three HAdVB3/7 recombinant strains, including two HAdVB66 strains (JN860676.1 and JX423386.1) collected from Argentina in 1987 and 2003, and strain NICED/23 − 01/1914 (OR039269.1) isolated from India in 2023, were also classified within Cluster 1. Other genotypes within species B were located in Cluster 2, Cluster 3 and Cluster 4. Most of the 29 outbreak strains were more closely related to HAdVB3 reference strains in evolutionary relationships, showing 99.2-99.9% nucleotide identity to strain SH20160055 (MW748667.1) collected in Shanghai, China in 2016. Six strains obtained in this study (SZZJ20240513, SZZJ20240515, SZZJ20240516, SZTC20241133, SZCS20241240 and SZCS20241242) exhibited higher nucleotide homology to HAdVB66 strain 87–922 (JN860676.1) and strain NICED/23 − 01/1914 (OR039269.1), with 98.5-99.0% and 98.4-99.6% nucleotide sequence identities, respectively (Fig. 2A).
The phylogenetic tree based on the hexon gene was also stratified into four cluster, consistent with the whole genome analysis. The intra-cluster evolutionary distances for Cluster 1, Cluster 3 and Cluster 4 were 0.049, 0.080 and 0.092, respectively. Among the 29 outbreak strains, two (SZZJ20240513 and SZTC20241133) clustered with HAdVB3 reference strains, with 98.9% nucleotide identity. In contrast, four strains (SZZJ20240515, SZZJ20240516, SZCS20241240 and SZCS20241242) were closely related to HAdVB7 reference strains, with 97.1-100% nucleotide identity. The remaining outbreak strains were nearly identical to HAdVB3 reference strains, with identities ranging from 99.9 to 100% (Fig. 2B).
The penton base gene sequences were categorized into three clusters, with the intra-cluster evolutionary distances ranging from 0.005 to 0.075, and the inter-cluster distances ranging from 0.159 to 0.208. Similar to the classification of the whole genome and hexon gene sequences, most outbreak strains belonged to Cluster 1 and were closely related to HAdVB3 reference strains. Notably, six strains (SZZJ20240513, SZZJ20240515, SZZJ20240516, SZTC20241133, SZCS20241240 and SZCS20241242), along with strain SZTC20241129, exhibited closer evolutionary relationships with HAdVB7 and HAdVB66 reference strains (Fig. 2C). The phylogenetic tree based on the fiber gene formed four clusters. In contrast to the trees based on the whole genome and the other two major capsid genes, all HAdVB7 reference strains formed a separate cluster along with several other genotypes within species B. All 29 outbreak strains clustered with epidemic HAdVB3 strains, sharing 98.2-100% nucleotide identity with HAdVB3 strains from neighboring regions, including strain SH20160055 (MW748667.1) from Shanghai in 2016 and strain ZJ20150111 (MW748671.1) from Hangzhou in 2015 (Fig. 2D).
Overall, most HAdV strains isolated from ILI outbreaks in Suzhou clustered closely with HAdVB3 reference strains. A subset showed genomic similarities to HAdVB7 or related recombinant genotypes, suggesting the presence of recombination events and warranting further investigation.
Amino acid variation analysis
The amino acid identities of the 29 outbreak strains were 95.2-100% for hexon, 99.4-100% for penton base, and 96.2-100% for fiber. The sequences obtained were highly consistent and nine representative strains were selected for amino acid variation analysis. Seven hypervariable regions (HVRs) on the surface of the hexon protein carry the type-specific antigenic determinants, which elicit protective antibodies and confer immunity against reinfection by homologous HAdV strains. HVR1 to HVR6 are situated within the loop 1 domain, and HVR7 is located in the loop 2 domain [27, 28]. Compared with the HAdVB3 reference strain collected from the United States in 2013 (OQ518322.1), the amino acid mutation N439D in loop 2 was shared by three samples (SZWJ20240505, SZZJ20240511, SZTC20241129), as well as in a previously reported Suzhou strain JSSZ1705_S31 from 2017. Four amino acid substitutions (H417N, A429T, N439A, M884L) in loop 2 were common in SZZJ20240513 and SZTC20241133, which were identical to the HAdVB3/7 recombinant strain NICED/23 − 01/1914 (OR039269.1). Consistent with the evolutionary relationships described in the phylogenetic tree of the hexon gene, the amino acid sequence in the loop 1 and loop 2 domains of SZZJ20240516 was identical to Beijing HAdVB7 strain isolated in 2017 (MT367401.1). Additionally, the substitutions in three remaining samples (SZZJ20240515, SZCS20241240, SZCS20241242) were inherited from either Beijing HAdVB7 strain (MT367401.1) or the recombinant strain NICED/23 − 01/1914 (OR039269.1) (Fig. 3A).
During the viral entry process, the fiber knob domain interacts with the primary cellular receptors (e.g., CAR, CD46 and DSG2), followed by interaction between the RGD loop of the penton base and secondary receptors such as αV integrins [3, 29]. Amino acid substitutions within these domains may alter receptor binding and influence viral infectivity. Compared with the HAdVB3 reference sequence (OQ518322.1), the substitution T462A in the penton base was present in nearly all strains obtained in the study except SZWJ20240505 and SZZJ20240511, as well as in Chinese reference strains isolated after 2013. Moreover, an additional substitution D325N in RGD loop was observed in three strains (SZZJ20240515, SZZJ20240516 and SZCS20241240) and mutation Q122R was observed in SZTC20241129. No additional mutations were found in SZZJ20240511. In contrast, a unique substitution, K171T, was detected in SZWJ20240505. This substitution is located adjacent to the HVR1 region (amino acids 149–169) and was absent in all reference strains (Fig. 3B).
In the fiber gene, the substitution T316R in the knob domain was shared by six outbreak strains. Among these, five strains (SZZJ20240513, SZZJ20240515, SZZJ20240516, SZCS20241240, SZCS20241242) carried two additional mutations (N22T, L23S) in the N-terminal domain, and one strain SZTC20241133 had an extra N72D mutation. The fiber sequences of SZWJ20240505 and SZZJ20240511 were identical to the HAdVB3 reference sequence (OQ518322.1). In contrast, the fiber sequence of SZTC20241129 was identical to those of two Shanghai HAdVB3 strains in 2011 (MK883603.1) and 2016 (MW748667.1), which were distinguished from OQ518322.1 solely by the N72D mutation (Fig. 3C).
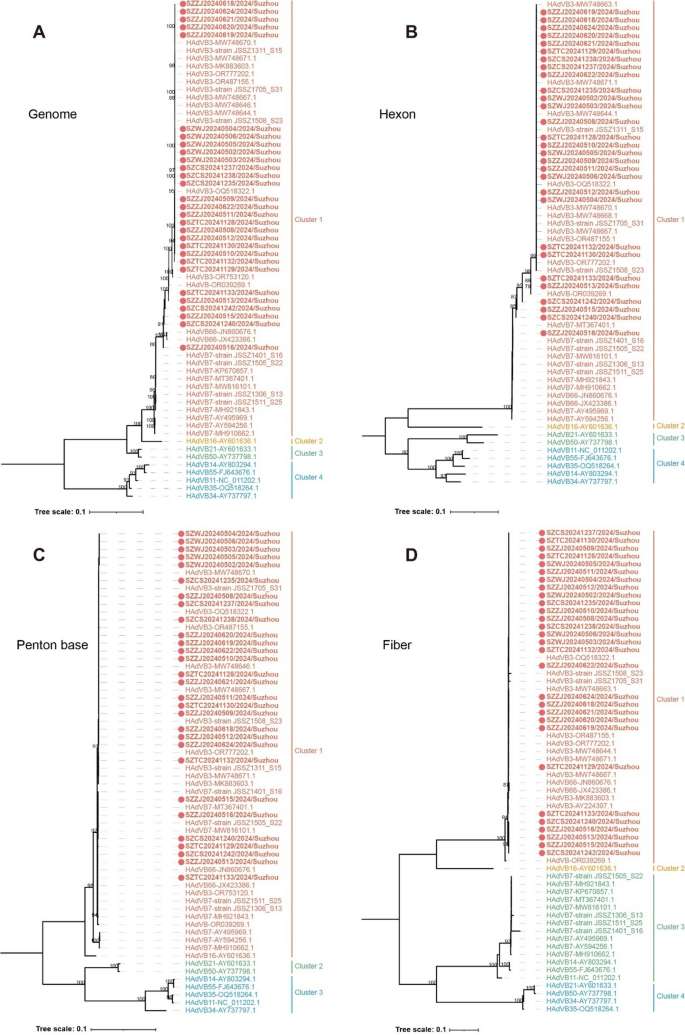
Phylogenetic analysis of HAdV strains isolated from 2024 ILI outbreaks in Suzhou. The trees for whole genome (A), hexon (B), penton base (C) and fiber gene (D) were constructed by the maximum likelihood (ML) method with IQ-TREE v2.3.6. The strains obtained in this study were highlighted in bold and marked with red circles ( ). Numbers above branches indicate bootstrap values, with those greater than 75% were shown
). Numbers above branches indicate bootstrap values, with those greater than 75% were shown

Amino acid variation analysis of hexon (A), penton base (B) and fiber (C) sequences obtained in this study. The loop 1 domain of hexon (amino acids 131–314) and knob domain of fiber (amino acids 124–319) were highlighted in pink, while the loop 2 domain of hexon (amino acids 405–454) and RGD loop of penton base (amino acids 299–362) were colored in yellow
Recombination analysis
Homologous recombination between HAdV types is a major driver of increasing viral genetic diversity and may influence infectivity and virulence. The recombination analysis of the 29 whole genome sequences revealed that seven strains (SZZJ20240513, SZZJ20240515, SZZJ20240516, SZTC20241129, SZTC20241133, SZCS20241240, SZCS20241242) were recombined from HAdVB3 and HAdVB7 genotypes, whereas the remaining strains were classified as HAdVB3 genotype without intertypic recombination (Supplementary Fig. 1). The recombination event of SZTC20241129 occurred with a beginning breakpoint at around 6,074 nucleotide location and an ending breakpoint at around 15,410 nucleotide location, encompassing partial E2B gene region, 52/55 kDa protein, protein IIIa precursor (pIIIa) as well as penton base, which recombined from the HAdVB7 strain isolated in Hubei, China in 2019 (MW816101) (Fig. 4A). In contrast to the recombinant pattern of SZTC20241129, the strain SZZJ20240516 harbored a HAdVB7-derived backbone and a HAdVB3-derived insertional fragment. The recombination breakpoints of SZZJ20240516 were comparable to those of SZZJ20240515, SZCS20241240 and SZCS20241242, which appeared with a recombinant region spanning L5-fiber, partial E3 and E4 gene regions derived from the HAdVB3 strain found in the United States in 2016 (OR777202.1) (Fig. 4B). The identified recombinant events of both SZTC20241129 and SZZJ20240516 were consistent in the RDP4 and SimPlot analysis. The average p values calculated across six of seven selected detection methods implemented in the RDP4 software were ranging from 7.41 × 10–130 to 2.49 × 10–33 for SZTC20241129 and 6.85 × 10–173 to 1.92 × 10–49 for SZZJ20240516, which further substantiated the occurrence of the recombination events (Supplementary Table 3).
The recombination pattern of SZZJ20240513 was similar to that of SZTC20241133, with two recombinant regions were detected by SimPlot analysis. The recombinant region 1 (around 18,257 − 25,219 nucleotide location of SZZJ20240513) encompassed partial protein VI precursor (pVI), hexon, DNA-binding protein (DBP) and partial 100 kDa hexon-assembly associated protein (100k) gene region, and the recombinant region 2 (around 27,927 − 33,987 nucleotide location of SZZJ20240513) covered partial E3 gene region, L5-fiber and partial sequences of E4 gene region. Both recombinant regions originated from an HAdVB7 strain isolated in the United States in 2007 (MH921843.1), while the backbone was derived from HAdVB3 (OR777202.1) (Fig. 4C). However, RDP4 analysis revealed that a contiguous gene region spanning both regions described above was derived from HAdVB3 (OR777202.1), with an internal fragment originating from HAdVB7 (MH921843.1) (Supplementary Table 3). To further validate the recombination events, phylogenetic analysis was performed for three gene regions, including the recombinant region 1 (Region 1, 18,257 − 25,218 nucleotide location of SZZJ20240513), recombinant region 2 (Region 2, 27,927 − 33,987 nucleotide location of SZZJ20240513) and the interjacent region (Region 3, 25,219 − 27,926 nucleotide location of SZZJ20240513). The ML trees indicated that both Region 1 and Region 2 clustered with HAdVB3 reference strains, whereas Region 3 clustered with HAdVB7 reference strains. The phylogenetic position supported the results of the recombination analysis (Fig. 5). Notably, the strain NICED/23 − 01/1914 (OR039269.1), collected from a hospitalized child during an outbreak in West Bengal, India between December 2022 and March 2023 [18], exhibited a similar recombination pattern to that of SZZJ20240513 (Fig. 4D). Importantly, such recombination patterns were not observed in HAdVB3 or HAdVB7 strains previously circulating in China (e.g., Suzhou, Zhejiang, Shanghai, Beijing, Guangzhou and Hubei) from 2011 to 2019, which were included in our earlier phylogenetic analysis.
Regarding the genotypes of the three major capsid proteins in the recombinant strains, three samples (SZZJ20240513, SZTC20241133, SZTC20241129) exhibited a P7H3F3 genetic constituent, whereas four samples (SZZJ20240515, SZZJ20240516, SZCS20241240, SZCS20241242) displayed a P7H7F3 genetic constituent, which were in agreement with the results of BLAST analysis.
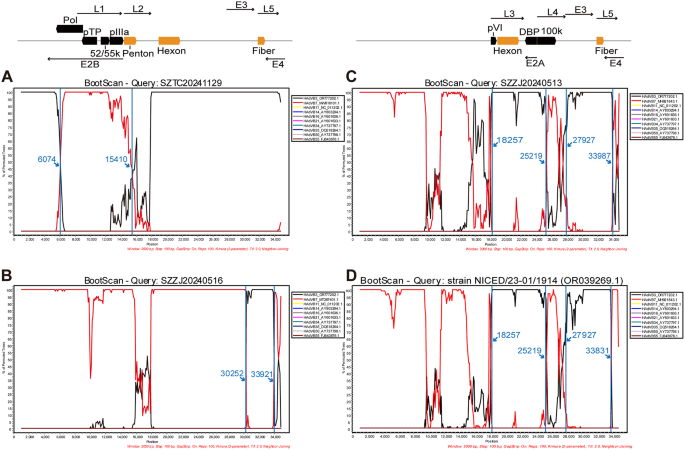
Genome recombination analysis of the three representative recombinant strains (A-C) and reference strain NICED/23 − 01/1914 (D) by SimPlot software. The 29 whole genome sequences obtained in this study shared high homology in nucleotide sequence and recombination pattern. Thus, the bootscan analysis results of three representative recombinant strains SZTC20241129 (A), SZZJ20240513 (B) and SZZJ20240516 (C) were shown and the recombination patterns of the remaining recombinant strains resembled any one of the three strains above
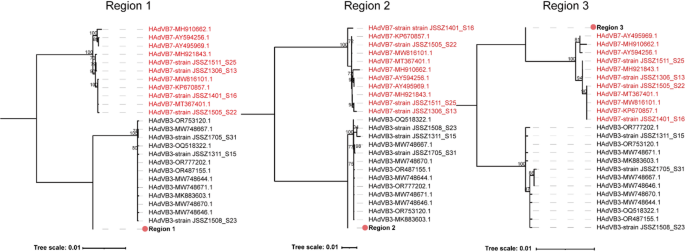
Phylogenetic analysis of potential recombinant regions of strain SZZJ20240513. The trees for the Region 1 (18,257 − 25,218 nucleotide location of SZZJ20240513), Region 2 (27,927 − 33,987 nucleotide location of SZZJ20240513) and Region 3 (25,219 − 27,926 nucleotide location of SZZJ20240513) were constructed by the maximum likelihood (ML) method using IQ-TREE v2.3.6. The HAdVB3 and HAdVB7 reference strains were colored in black and red, respectively. The corresponding fragments in the strain SZZJ20240513 was highlighted in bold and labeled with the red solid circle ( ). Numbers above branches indicate bootstrap values, with those greater than 70% were shown
). Numbers above branches indicate bootstrap values, with those greater than 70% were shown