QUT researchers have revealed key biological processes that enable corals to attach to reef surfaces, a discovery with strong potential to improve coral restoration worldwide.
Published in Royal Society Open Science, the research led by Dr. Brett…
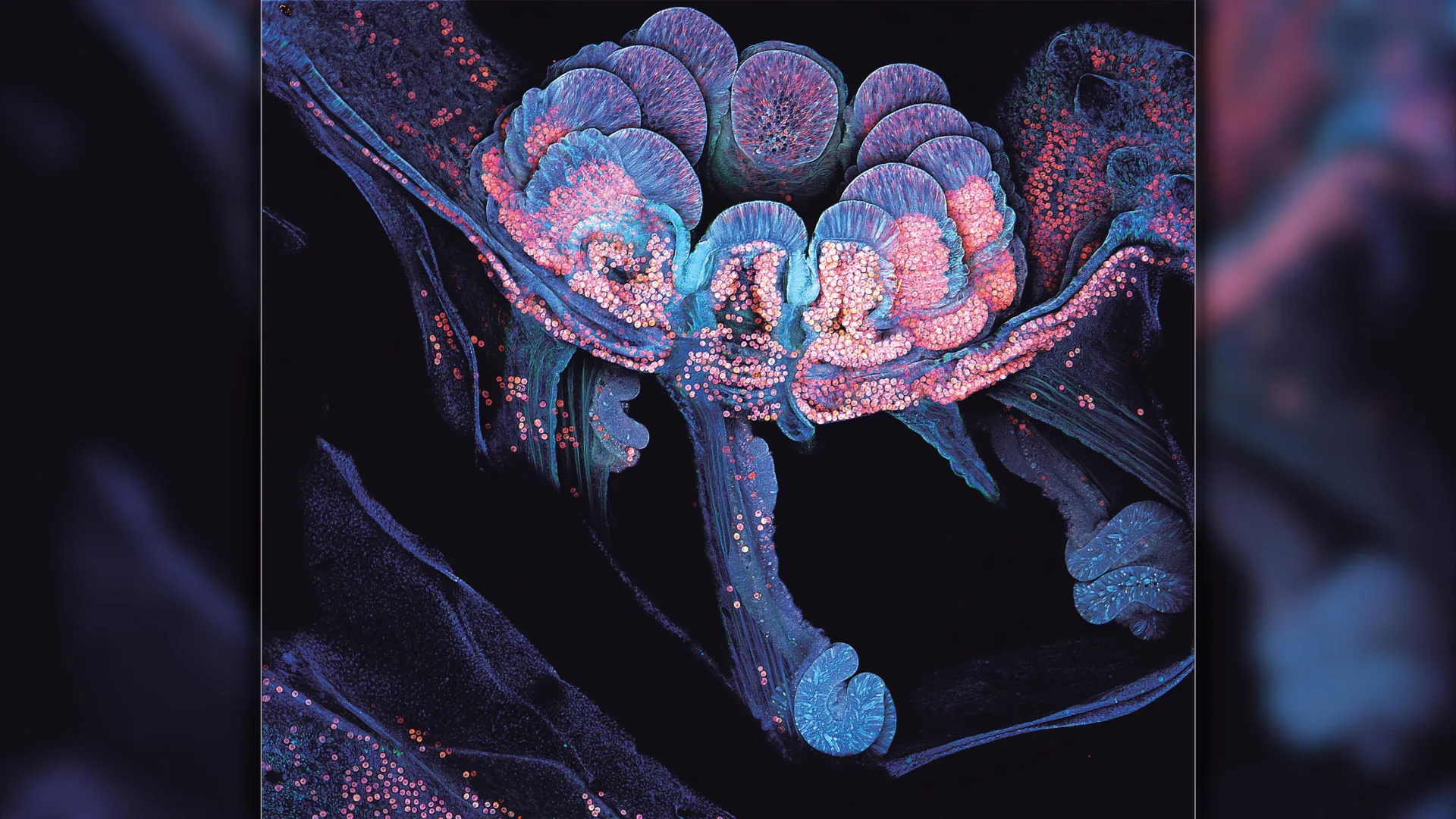
QUT researchers have revealed key biological processes that enable corals to attach to reef surfaces, a discovery with strong potential to improve coral restoration worldwide.
Published in Royal Society Open Science, the research led by Dr. Brett…