Study design
This is a 1:1 randomized, controlled, parallel-group, single-center, single-blind superiority trial, which aims to evaluate the benefits of integrated blood purification therapy in SA-AKI patients identified by CCL14 [21]. The primary objective of this trial is to evaluate whether HBP therapy combining CRRT with hemoperfusion can reduce the 30-day all-cause mortality in SA-AKI patients identified by CCL14, compared with the control group receiving CRRT alone, so as to provide high-level evidence for determining the optimal treatment approach for SA-AKI patients in clinical practice. The secondary objectives are to clarify whether the HBP mode affects the 90-day all-cause mortality, the incidence of progression to chronic kidney disease (CKD), and other relevant clinical indicators of SA-AKI patients identified by CCL14.
This randomized controlled trial protocol follows the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 statement and Consolidated Standards of Reporting Trials (CONSORT) 2010 (Additional file 1–2) [22].
Patient and public involvement
Patients or the public were not involved in the design, implementation, reporting, or dissemination plans of our study.
Ethics approval and registration
This clinical trial was approved by the Medical Ethics Committee of Northern Jiangsu People’s Hospital in April 2024 (Approval Number: 2024ky081). This study was registered in the Chinese Clinical Trial Registry (CHICTR) in December 2024 (Registration Number: ChiCTR2400093572).
Protocol amendments
All modifications to the study protocol will be submitted to the Medical Ethics Committee of Northern Jiangsu People’s Hospital for review and approval. Researchers shall not make any modifications to the protocol until they receive the approval reply from the Ethics Committee.
Consent or assent
Given that critically ill patients may have impaired cognitive abilities, researchers in charge of patient enrollment will provide written informed consent forms to the subjects, their relatives, or legal representatives, and inform them of the purpose, interventions, benefits, and risks of this trial. Interventions, data collection, and sample collection will all be implemented after obtaining written informed consent.
Confidentiality
This trial strictly protects the privacy and personal information of subjects through an anonymization strategy. All subjects’ names and hospital admission numbers will be replaced with unique study-specific serial numbers after enrollment. All paper documents and electronic data containing personal information will be restricted in access, with access permissions granted only to designated researchers. During the data analysis process, researchers will use anonymized data for analysis. Without obtaining the subject’s written consent, the subject’s research information shall not be used for purposes other than clinical research.
Inclusion and exclusion criteria
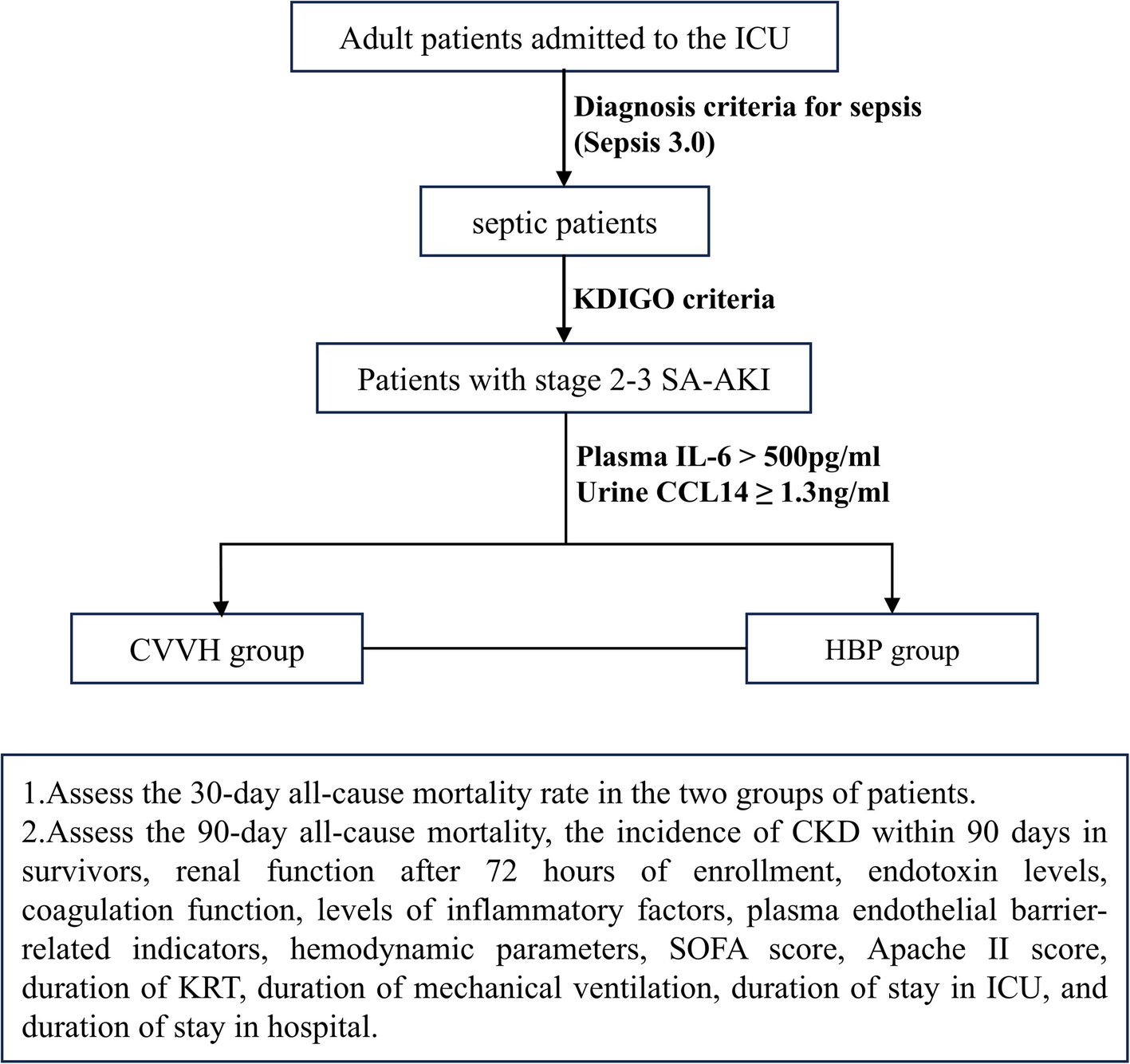
This study will screen patients admitted to the Department of Critical Care Medicine of Northern Jiangsu People’s Hospital affiliated to Yangzhou University from December 10, 2024, to December 31, 2025, and eligible patients meeting the following criteria will be included in the study: Adult patients admitted to the ICU, aged 18 years or older; meeting diagnosis criteria for sepsis (Sepsis 3.0) [1]; diagnosed with stage 2–3 AKI according to the KDIGO criteria within 1–7 days after sepsis diagnosis [23]; meeting the indications for CRRT [24]; plasma IL-6 > 500 pg/ml; urine CCL14 ≥ 1.3 ng/ml [25]; and providing informed consent. The diagnosis criteria for sepsis (Sepsis 3.0) are shown in Table 1. The indications for CRRT are shown in Table 2. The staging of acute kidney injury according to KDIGO 2012 guidelines is shown in Table 3.
Patients meeting the following criteria were excluded: those with allergies to resins, heparin, or fish protein; patients with pre-existing stage 4–5 CKD; patients who received kidney replacement therapy (KRT) before entering the ICU; anuric patients; those in the terminal stage; those who opted for withdrawal of treatment during the study period; patients who died within 72 h after entering the ICU.
The baseline serum creatinine level is defined as the median value of serum creatinine measured from 6 months to 6 days prior to admission. If no serum creatinine data is available before enrollment, assume a baseline glomerular filtration rate (GFR) of 75 mL/min/1.73 m2 and estimate the baseline serum creatinine value using the Modification of Diet in Renal Disease (MDRD) equation for estimating Glomerular Filtration Rate (eGFR = 186 × (SCr)−1.154 × (age)−0.203(× 0.742 if female)) [26]. For patients with CKD stages 1–3, if no serum creatinine data was available before enrollment, the first serum creatinine value after admission was used as the baseline creatinine. Stage 4–5 CKD is defined as a GFR < 30 mL/min/1.73 m2, sustained for more than 3 months [27].
Randomization and concealment mechanism
In this trial, block randomization was employed to allocate participants, ensuring comparable baseline characteristics between the experimental and control groups. The procedure was as follows: An independent researcher generated a 1:1 randomized allocation sequence using computer software. Variable block sizes were used to minimize predictability of the randomization schedule. Within each block, participants were allocated to the experimental group (HBP group) and the control group (CVVH group) at a 1:1 ratio to maintain approximate balance in sample sizes between the two groups across different stages of the study. After the randomization sequence is generated, an independent researcher responsible for allocation concealment will seal the group assignment information in opaque envelopes. Each envelope will be marked solely with a unique participant identifier corresponding to one subject. All envelopes will be stored by the independent researcher and opened in sequential order by a designated individual only after the participant has met the inclusion criteria and signed the informed consent form.
Blinding
In this trial, blinding is implemented for the subjects, and researchers and medical staff do not inform the subjects of their group allocation. Due to the significant differences in operational procedures and equipment setups between the experimental group (HBP group) and the control group, it is impossible to blind the researchers, doctors, and nurses. Owing to the limited number of research personnel, this trial does not implement blinding for the research implementers and data analysts. In the event of adverse events or changes in intervention measures, the principal investigator will report to the ethics committee and the funding institution.
Intervention
The enrolled patients were randomly allocated into the CVVH group and the HBP group. The intervention strategy for the HBP group was to use the 2–1–1 treatment protocol with the HA380 blood perfusion filter (Jafron, China) during CRRT treatment using the CVVH mode with AN69 ST100 blood filter (Baxter, USA) [28]. Within 24 h after enrollment, 2 perfusion filters were used, followed by 1 perfusion filter each day for the next 2 days, with each HA380 blood perfusion filter being used for 6 h. If the subject shows significant improvement within 72 h after undergoing HBP, the treatment with the HA380 blood perfusion filter will be terminated, and they will be included in the late-stage subgroup analysis. The intervention strategy for the CVVH group is to use the AN69 ST100 blood filter (Baxter, USA) to choose the CVVH mode for CRRT (Fig. 1). All enrolled patients are prohibited from using other hemoperfusion devices or hemofilters with the function of adsorbing inflammatory factors during the trial. For the relevant concomitant care and interventions, the treatment recommendations in the disease guidelines shall be followed.
Flowchart of study protocol
Strategies for handling protocol non-adherence
All non-compliance events will be recorded in detail by the investigator. For reversible non-compliance events, the research staff will make every effort to correct them and document the correction results. If situations arise that endanger the safety of the subject or if persistent non-compliance renders the data unusable for outcome assessment, the principal investigator may decide to terminate the subject’s participation in the trial. In statistical analysis, the intention-to-treat (ITT) principle will be adopted to avoid bias caused by excluding non-compliant subjects. For missing data resulting from non-compliance, the multiple imputation method will be used for data supplementation.
Study primary outcomes
The primary outcome is all-cause mortality rate 30 days after enrollment.
Study secondary outcomes
The secondary outcomes include: 90-day all-cause mortality rate, incidence of CKD within 90 days in survivors, changes in kidney function (serum creatinine, 24-h urine volume) after 72 h of enrollment, changes in endotoxin levels, changes in coagulation function (PT, INR, APTT, D-Dimer, FG, TT), changes in inflammatory factor levels (12 cytokines), changes in plasma vascular endothelial barrier-related indicators (syndecan-1, E-selectin, Ang-2, VE-cadherin, claudin-5), changes in hemodynamic parameters (mean arterial pressure, MAP, central venous pressure, CVP, dose of norepinephrine), changes in SOFA score, changes in Apache II score, duration of kidney replacement therapy (KRT) during hospitalization, duration of mechanical ventilation, duration of stay in ICU, and duration of stay in hospital.
Participant timeline
Baseline data collected at enrollment include: demographic data (age, gender, body mass index), AKI staging (according to KDIGO criteria), underlying diseases (hypertension, diabetes, chronic obstructive pulmonary disease, cardiovascular diseases, liver diseases), complications (cerebral edema, respiratory failure, heart failure, acute liver injury, gastrointestinal bleeding, anemia, hypoalbuminemia), respiratory support (including pulse oximetry saturation, PaO2/FiO2, type of mechanical ventilation), hemodynamic parameters (MAP, CVP, dose of norepinephrine), SOFA score, and Apache II score.
Laboratory data includes: kidney function (serum creatinine, 24-h urine volume), endotoxin levels, coagulation function (PT, INR, APTT, D-Dimer, FG, TT), inflammatory factor levels (12 cytokines), and plasma vascular endothelial barrier-related indicators (Syndecan-1, E-selectin, Ang-2, VE-cadherin, Claudin-5).
Sepsis infection status data includes: site of infection (lungs, abdomen, skin, urinary tract, bloodstream, nervous system, heart valves and myocardium, unknown site, other sites), culture results (Gram-positive bacteria, Gram-negative bacteria, Gram-positive and negative bacteria, fungi, viruses, no results), types of pathogens (Staphylococcus aureus, Pseudomonas aeruginosa, Acinetobacter baumannii, Escherichia coli, Klebsiella pneumoniae, Candida albicans, other pathogens).
Blood and urine collection and testing: At the time of enrollment and 72 h after enrollment, 10 ml of blood and urine samples are collected from the subjects, which are then centrifuged at 3000 rpm for 5 min, left to stand for 30 min, and the supernatant is then stored in an environment of ≤ −70 °C for testing. We use enzyme-linked immunosorbent assay to detect urine CCL14 concentration, plasma vascular endothelial barrier-related indicators (Syndecan-1, E-selectin, Ang-2, VE-cadherin, Claudin-5), and quantitative detection of endotoxin content in plasma using the Limulus Amebocyte Lysate (LAL) assay endpoint chromogenic method. During the detection process, the operators are not informed about the experimental research content and strictly follow the instructions for the reagent detection. Information on the time points of trial enrollment, intervention, and assessment is shown in Table 4.
Data management
The researchers used Case Report Form (CRF) for data collection, registered and managed data on the ResMan website.
Data monitoring
Since this trial is a single-center study, data monitoring will be conducted by members of the research team led by the principal investigator, and a data monitoring committee is not required.
Members of the research team will continuously monitor the trial process, clinical data, and sample collection. If patients in the HBP group experience significant adverse events, such as a significant increase in CRRT anticoagulant dosage or deterioration of coagulation indicators, or if early results indicate obvious flaws in the CVVH group, the principal investigator will decide to terminate this trial.
Sample size calculation
This study is a randomized controlled trial design, with the HBP group as the experimental group and the CVVH group as the control group. The primary evaluation index observed is the 30-day all-cause mortality rate of the study subjects. According to the literature review results, the 30-day all-cause mortality rate (p1) of the experimental group in previous related studies is 0.71, and the 30-day all-cause mortality rate (p2) of the control group is 0.82. With a two-sided α = 0.05 (one-sided α = 0.025), a power of 1-β = 0.9, a sample size ratio of 1:1 between the experimental and control groups, and a margin of clinical superiority set at 0.1, the sample size calculated using R software is 85 for the experimental group and 85 for the control group. Taking into account a 10% loss to follow-up and refusal rate, a minimum of 95 participants is required for both the experimental and control groups. This results in a total of 190 participants included in the study [29]. Researchers monitor the patients admitted to the ICU in real-time and conduct screening to ensure an adequate number of enrolled patients.
Statistical analysis
This study utilized Case Report Forms (CRFs) for data collection, ResMan for data management, and R 4.3.1 and R Studio for data analysis. Normally distributed continuous data are presented as mean ± standard deviation (i ± s), while non-normally distributed continuous data are presented as median (interquartile range) M (P:25, P:75), and categorical data are presented as frequency and percentage. For comparing two groups of quantitative data, Wilcoxon test or t-test are used. For comparing two groups of qualitative data, chi-squared test or Fisher’s exact test is used. Survival analysis is conducted using Kaplan–Meier curve, and survival rate comparison is conducted using log-rank test. A p-value < 0.05 indicates a statistically significant difference. Use the method of deleting missing values or multiple imputation to handle the problem of missing data.
In this study, no interim analysis of the outcome endpoints will be conducted until the completion of the study, as well as the enrollment of all subjects and the completion of their follow-up.
Adverse events
Adverse events (AEs) in this study include: bleeding, thrombosis, infection, and complications related to hemoperfusion and CRRT treatment (i.e., hypotension, internal environment disturbance). Researchers responsible for adverse events will record these events within 3 days after enrollment. Serious adverse events (SAEs) include events that result in death, are life-threatening, require hospitalization or prolongation of hospital stay, or cause permanent or severe disability or dysfunction. Researchers responsible for adverse events will follow up on serious adverse events (SAEs) within 90 days after enrollment, or until the events are resolved, and submit a written report to the ethics committee. After an adverse event occurs, researchers and physicians will develop a standardized treatment plan and conduct long-term follow-up until the patient’s condition improves or 90 days after the event. Since the HA380 disposable hemoperfusion cartridge is a tested and approved medical device, no harm will be directly caused by the intervention. In the event that a subject suffers previously unknown harm directly caused by the trial intervention, the medical expenses will be covered by the trial funds, and corresponding compensation will be made in accordance with relevant laws and regulations.
Auditing
Beyond the monitoring and reporting of adverse events and harmful complications to patients, this study does not require specific measures for auditing the trial implementation.