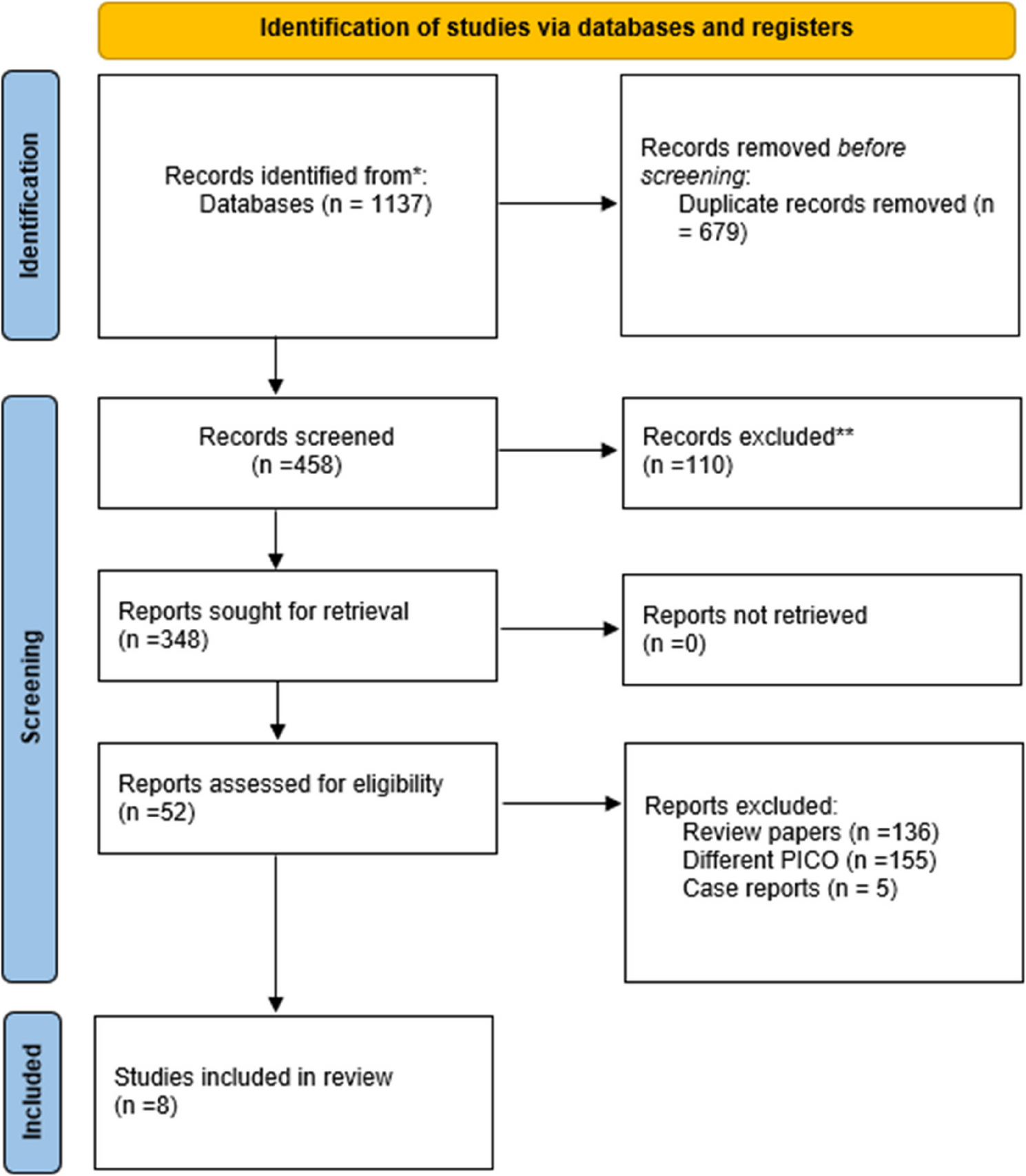
The relationship between the presence of H. Pylori bacteria and the likelihood of developing AAA has drawn considerable attention, given its potential implications for preventive and therapeutic measures. Although various studies have explored this connection, their findings remain inconsistent. To address this gap in knowledge, we conducted a systematic review and meta-analysis of existing literature involving, aiming to provide a comprehensive and definitive assessment of the relationship between H. pylori and AAA. This is the first study to synthesize the existing literature on this topic, and our findings offer the highest level of evidence on this association.
The rates of AAA in patients with HPI varied significantly across the seven selected studies, according to Ziver et al. [9]who reported rates as high as 67%, while Jones et al. [22] reported rates as low as 22%. Despite this discrepancy, the overall calculated event rate across all studies was 41%. The reasons for this variation could be attributed to disparities in study design, population, and inclusion and exclusion criteria. The data analysed to assess the RR of the association between HPI and AAA revealed a significant risk, but due to high heterogeneity, three studies by Cheng et al., Bouhoutsos et al., and Ziver et al. were excluded from analysis [9, 14, 23]. The remaining studies by showed a significant increased risk of AAA associated with HPI [22, 24, 25]. Although there was a reduction in heterogeneity, the test for overall effect was not statistically significant, even after excluding the heterogeneous studies. In our sensitivity analysis, the exclusion of studies identified as major contributors to heterogeneity resulted in a reduction of the pooled RR from 1.54 to 1.17, with the loss of statistical significance (p = 0.14). This finding indicates that the initial significant association between H. pylori infection and AAA may have been driven largely by a small number of heterogeneous studies, each with distinct methodological and population characteristics. Consequently, these results should be interpreted with caution. While the adjusted estimate demonstrates improved statistical homogeneity (I² = 0%), the diminished effect size suggests that further large-scale, methodologically consistent studies are necessary to confirm or refute this association. This reinforces the importance of future research employing standardized diagnostic criteria for H. pylori, uniform definitions of AAA, and consistent adjustment for confounding factors.
Chronic inflammation, a hallmark of H. pylori infection, could potentially worsen and cause AAA. This bacterium provokes systemic inflammatory responses, which may compromise the integrity of arterial walls and lead to AAA. Moreover, H. pylori may directly infect existing aortic aneurysms, indicating an infectious component in aneurysm progression. The interaction between mast cells (MCs) also exacerbates vascular damage by triggering additional inflammation and endothelial dysfunction, thereby heightening the cardiovascular risks associated with AAA [26,27,28].
Further research into the relationship between HPI and AAA is warranted. The pathogenicity factors of H. pylori with CagA + strains need to be explored, as literature consistently indicates their capacity to damage the gastric mucosa compared to CagA- strains and their involvement in exacerbating the inflammatory response associated with AAA. The proposed pathogenesis of H. pylori involves the elicitation of vacuolation of the gastric epithelium by a high-molecular-weight toxin, leading to an enhanced local inflammatory response. The immunodominant protein associated with this toxin is the cytotoxin-associated gene A (cagA). A strong correlation between HPI and atherosclerosis has been established, with CagA positivity being linked to a more significant relationship. This relationship is demonstrated by increased carotid intima-media thickness (CIMT), impaired flow-mediated dilation (FMD), and elevated pulse wave velocity (PWV) [29, 30].
Increased levels of HPI in patients with CagA positivity have been found to significantly increase the risk of developing atherosclerotic cardiovascular diseases. This relationship is supported by the presence of anti-CagA antibody titers in patients with unstable angina and the detection of CagA antigens in coronary atherosclerotic plaques [31]. Furthermore, individuals with H. pylori infection, particularly those with CagA seropositive strains [32]are more likely to experience adverse cardiovascular events, such as myocardial infarction and cerebrovascular disease [33, 34]. Given the similarities between atherosclerosis and AAA in terms of their pathophysiology, biomarkers, and genetics, it is reasonable to expect a link between AAA and HPI [35, 36]. According to a study, H pylori DNA was discovered in the carotid atherosclerotic plaques of 20 patients, amounting to 53% of the total [37]. This is a significant finding, considering that H pylori is an infectious agent that typically invades the gastrointestinal tract directly. However, recent information and ongoing research on the implications of infectious processes and the possibility of direct arterial invasion suggest that this discovery may not be as far-fetched as previously thought [37]. According to a study conducted by Wu et al., a small sample of 168 patients with carotid atherosclerosis and infected with the H. pylori CagA + strain was found to have a thicker carotid intima-media, which increased the likelihood of having unstable plaques. Additionally, there was an overexpression of the YKL-40 marker in these patients. Compared to other commonly used markers, such as C-reactive, the YKL-40 marker was found to be a more effective predictor of plaque status and carotid atherosclerosis outcomes in CagA-positive HPI patients. The similar pathogenesis of atherosclerosis and AAA further supports our hypothesis regarding the link between HPI CagA + and AAA [38].
This meta-analysis provides a substantial input for understanding the relationship between HPI and AAA, presenting detailed findings even in the face of methodological difficulties. The data collected and analysed show a clear connection between HPI and a higher risk of developing AAA, which is supported by the calculated event rate of 0.414 across various studies. However, the high degree of heterogeneity observed in the studies, resulting from differences in study design, population, and inclusion criteria, necessitated the removal of some studies to achieve a clearer understanding, even though the overall effect remained statistically insignificant. This implies that while chronic inflammation caused by HPI, particularly the CagA + strains, may be involved in atherosclerotic processes similar to those observed in AAA, it is difficult to draw definitive conclusions about the cause and mechanisms involved. The findings emphasize the need for more rigorous research to further explore this relationship.
Given that this meta-analysis relies on observational studies, it is subject to a number of limitations that could impact the validity of its conclusions. One of the main challenges is the inconsistent control of confounding variables across studies, which could result in biased findings. Additionally, these studies often employ varied methodologies, populations, and outcomes, which can complicate data aggregation and interpretation [39]. The initial analysis used studies that displayed significant heterogeneity, with differences in geographical and temporal contexts, study designs, and populations. In addition to the limitations already discussed, our findings should be interpreted in light of several further considerations. First, the possibility of publication bias cannot be excluded, as studies with null or negative results may be underrepresented in the published literature, potentially inflating the observed association. Second, unmeasured confounding variables—such as socioeconomic status, dietary habits, or co-existing inflammatory conditions—may have influenced the reported effect sizes, especially given that many included studies were observational in nature and differed in their approach to adjustment. Third, the heterogeneity in study populations, diagnostic methods for H. pylori, and definitions of AAA across the included studies may limit the generalizability of our findings to all settings. These factors highlight the difficulties in synthesizing and interpreting the broader implications of these studies on the relationship between AAA and HPI, given the diversity in methodologies and targeted populations.