Installation of BLAST, clustal omega and ruby
BLAST (blastn and makeblastdb version 2.8.1+) was downloaded from the NCBI website https://blast.ncbi.nlm.nih.gov/doc/blast-help/downloadblastdata.html. It was then stored in the /blast_directory/bin directory created on the local computer. Clustal Omega was downloaded from the Clustal Omega website http://www.clustal.org/omega/#Download. Ruby was downloaded from the Ruby website https://www.ruby-lang.org/en/downloads/. They were installed as instructed in each web site.
16S rRNA nucleotide sequences
The 16 S rRNA nucleotide sequences were obtained from the nucleotide database in GenBank. In GenBank, many 16 S rRNA entries have only partial sequences registered. To obtain full-length sequences, we first searched GenBank using queries that included the bacterial name and the word “complete” in the title, and “16S” anywhere in the entry. We then extracted the 16 S rRNA regions from GenBank records in which the FEATURE field was annotated as rRNA with the product labeled “16S ribosomal RNA.” These steps were repeated, and we retrieved up to the first 1000 sequences. For M. gordonae, which had only a small number of entries, we used sequences derived from whole genome sequencing data along with all GenBank rRNA entries for M. gordonae that were longer than 1400 bp. In summary, we obtained a total of 20,309 copies of the 16 S rRNA from 41 species of bacteria.
16S rRNA consensus sequence
The 16 S rRNA sequences that belong to a bacterial species were aligned using Clustal Omega to obtain the consensus sequence for that species. Subsequently, the consensus sequences for all bacterial species were aligned using Clustal Omega. We designed PCR primers based on the aligned sequences.
Match rate between PCR primers and 16S rRNA sequences
The match between either of the PCR primers and the 16 S rRNA sequence was measured using the Levenshtein distance. A Levenshtein distance of 0 indicates a perfect match, 1 indicates a single mismatch, and 2 or more indicates a mismatch of 2 or more bases.
Local database of pneumonia-causing bacteria
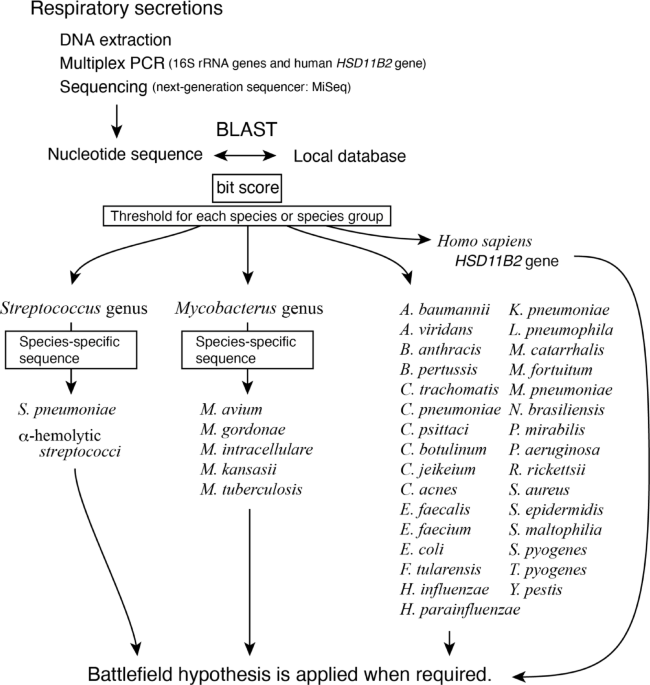
The sequences flanked by PCR primers from the consensus sequences of each bacterial species were collected and used to create a local database of pneumonia-causing bacteria. In addition, we also registered the consensus sequences of four α-hemolytic streptococci (S. mitis, S. oralis, S. salivarius, and S. sanguinis) to differentiate S. pneumoniae from them, and the sequence of the human HSD11B2 gene for the battlefield hypothesis. The specificity of the S. pneumoniae-specific nucleotide sequence was further confirmed in six additional species of α-hemolytic streptococci (S. anginosus, S. constellatus, S. gordonii, S. intermedius, S. parasanguinis, and S. vestibularis).
Bacterial and human genomic DNA
The genomic DNA of Escherichia coli (12713G), Pseudomonas aeruginosa (106052G), and Klebsiella pneumoniae subsp. pneumoniae (14940G) were purchased from the Biological Resource Center, NITE (Chiba, Japan). The genomic DNA of Staphylococcus aureus subsp. aureus Rosenbach ATCC 700699D-5, Streptococcus pneumoniae (Klein) Chester ATCC BAA-255D-5, Moraxella catarrhalis (Frosch and Kolle) Bovre ATCC 25240D-5, Haemophilus influenzae (Lehmann and Neumann) Winslow et al. ATCC 51907D, Proteus mirabilis Hauser ATCC 12453D, Acinetobacter baumannii ATCC BAA-1605D-5, and Mycobacterium tuberculosis (Zopf) Lehmann and Neumann ATCC 27294D-2, were purchased from the American Type Culture Collection (Rockville, MD, USA). Human placental genomic DNA was purchased from Promega Corp. (Madison, WI, USA).
Nucleotide sequencing by a next-generation sequencer
Two sets of primer mixes that simultaneously amplify 16S rRNAs and human HSD11B2 gene were prepared. The test solution was divided into two portions, PCR-amplified with both primer mixes, and then combined. Each reaction contained a primer mix (250 nM each), 1× PrimeSTAR GXL Buffer (Takara Bio Inc., Kyoto, Japan), 200 nM dNTPs, and 0.625 U PrimeSTAR GXL DNA Polymerase in a final volume of 25 µL. The cycling parameters were 94 °C for 120 s, followed by 30 cycles of 94 °C for 10 s, 55 °C for 15 s, and 68 °C for 35 s. The amplified products were purified using the Agencourt AMPure XP system (Beckman Coulter, CA, USA), and the nucleotide sequences were determined as described in a previous report (Inoue, Shiihara et al. 2017) using the MiSeq Reagent Kit V3 (Illumina). In that report, a second PCR was performed with primers that included an adapter sequence for loading onto the MiSeq, followed by a third PCR with indexed primers to distinguish multiple samples.
Cheryblast + ob analysis
Cheryblast + ob (check sequence query with BLAST + for bacteria: Deposited in https://doi.org/10.5281/zenodo.14759736) is a wrapper program for BLAST that we developed, incorporating a novel algorithm of our own design. It uses BLAST for the nucleotide homology analysis. The FASTQ files output from the MiSeq were analysed using Cheryblast + ob. Each read was classified into 37 types of pneumonia-causing bacteria 16 S rRNA, 4 α-hemolytic streptococci 16S rRNAs, the human HSD11B2 gene, or sequences that did not match any of these categories.
Effect of sequencing errors
To investigate the impact of sequencing errors on the classification of 16 S rRNA, we introduced mutations at rates ranging from 0 bp/500 bp to 5 bp/500 bp into the 16S rRNA sequences downloaded from GenBank. We then identified bacterial species using Cheryblast + ob. This process was repeated 100 times to calculate the average sensitivity and specificity for the identification of bacterial species.
A simulated reaction using DNA mixtures mimicking airway secretions
A two-fold dilution series of genomic DNA was created, starting with 65,000 copies of DNA derived from A. baumannii, E. coli, H. influenzae, K. pneumoniae, M. tuberculosis, M. catarrhalis, P. aeruginosa, P. mirabilis, S. aureus, S. pneumoniae, or human DNA. We set 65,000 copies as the target and used 32,500 copies, 16,250 copies, 8,125 copies, 4,063 copies, and 2,032 copies as competitors. Samples were prepared by arbitrarily selecting and mixing one target and one competitor of 32,500 copies, one competitor of 16,250 copies, and so on. These samples were then amplified using the PCR primers set in this study, sequenced with MiSeq, and analysed with Cheryblast + ob.