The New York Watch Auction: XIII, which included the sale of filmmaker Francis Ford Coppola’s custom watch, became the highest-grossing watch auction in history on Sunday.
The auction’s 10th anniversary for Phillips in association with…

The New York Watch Auction: XIII, which included the sale of filmmaker Francis Ford Coppola’s custom watch, became the highest-grossing watch auction in history on Sunday.
The auction’s 10th anniversary for Phillips in association with…

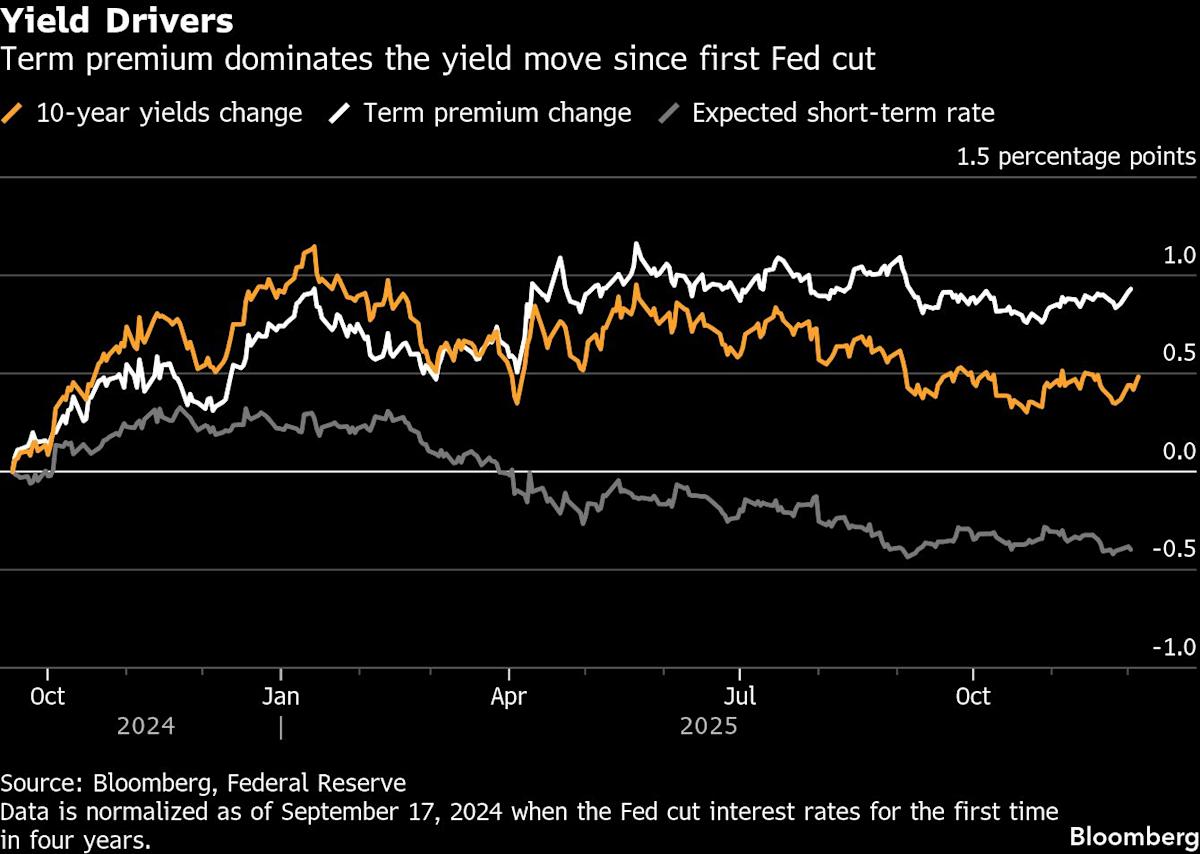
(Bloomberg) — The bond market’s reaction to the Federal Reserve’s interest-rate cuts has been highly unusual. By some measures, a disconnect like this, with Treasury yields climbing as the central bank lowers rates, hasn’t been seen since the 1990s.
What the divergence indicates is a matter of heated debate. Opinions are all over the place, from the bullish (a sign of confidence that recession will be averted) to the more neutral (a return to pre-2008 market norms) to the favorite culprit of the so-called bond vigilantes (investors are losing confidence the US will ever rein in the constantly swelling national debt).
Most Read from Bloomberg
But one thing is clear: the bond market isn’t buying President Donald Trump’s idea that faster rate cuts will send bond yields sliding down and, in turn, slash the rates on mortgages, credit cards and other types of loans.
With Trump soon able to replace Chair Jerome Powell with his own nominee, on top of everything else is the risk of the Fed squandering its credibility by caving to political pressure to ease policy more aggressively — which could backfire by fanning already elevated inflation and pushing yields higher.
“Trump 2.0 is all about getting long-term yields down,” Steven Barrow, head of G10 strategy at Standard Bank in London. “Putting a political figure at the Fed will not get bond yields down.”
The Fed started pulling its benchmark rate down from a more than two-decade high in September 2024 and has since cut it by 1.5 percentage points to a range of 3.75% to 4%. Traders see another quarter point cut after the next meeting on Wednesday as virtually assured and are pricing in two more such moves next year, which would bring its rate to around 3%.
Yet, key Treasury yields — which serve as the main baseline for the borrowing costs paid by American consumers and corporations — haven’t come down at all. Ten-year yields have risen nearly half a percentage point to 4.1% since the Fed started easing policy and 30-year yields are up over 0.8 percentage point.
Normally, when the Fed moves short-term policy rates up and down, long-term bond yields tend to follow. Even in the only two easing cycles outside of recessions over the past four decades – in 1995 and 1998, when the Fed cut only 75 basis points each time — the 10-year yield dropped outright or rose less than they have during the current episode.

Ted Sarandos scored a critical sit-down with President Donald Trump in the weeks leading up to Netflix‘s successful $82.7 billion agreement to buy Warner Bros. and HBO Max, according to sources familiar with the confab. The meeting, which…

All three dose groups (50 mg, 100 mg and 200 mg) showed impressive monotherapy efficacy, with VGPR+ (very good partial response or better) of ≥70% despite limited follow-up; evidence shows that these responses are expected to deepen over time
Across all dose groups, 95% (19 of 20 patients) of all evaluable VGPR+ patients achieved minimal residual disease negative status
Data featured in an ASH oral presentation; LINKER-MM4 is the first clinical trial to evaluate a BCMAxCD3 bispecific monotherapy in NDMM and is part of a broad clinical development program evaluating Lynozyfic-based regimens in earlier lines of treatment
Regeneron to host virtual ‘Regeneron Roundtable’ investor event to discuss its multiple myeloma development program on Wednesday, December 10 at 8:30 a.m. ET
“The treatment of newly diagnosed multiple myeloma often relies on complicated combinations of quadruplet or triplet regimens, each with its own toxicities, in order to achieve rapid and durable responses, which can be incredibly burdensome for these patients,” said
LINKER-MM4 is an ongoing, open-label Phase 1/2 trial investigating Lynozyfic in adults with NDMM. During a Phase 1A (dose escalation) cohort, patients were treated with a step-up dosing regimen followed by 50 mg, 100 mg or 200 mg doses of Lynozyfic. The lowest (50 mg) and highest (200 mg) tolerated doses were selected for further evaluation in the Phase 1B (dose-expansion) cohort. Among the 45 treated patients in both Phase 1A and 1B, 28 were transplant eligible, and 17 were transplant ineligible.
Across all dose levels (n=45), there was a 1.2 months median time to onset of response (range: 1-4.5 months). All three dose groups (50 mg, 100 mg and 200 mg) showed impressive efficacy, with a VGPR+ (very good partial response or better) of ≥70% with limited follow-up. Evidence shows that these responses are expected to deepen over time. Across all dose groups, 95% (19 of 20 patients) of all minimum residual disease (MRD) evaluable VGPR+ patients achieved MRD negative status at 10-5 sensitivity.
Across all dose levels, the most common treatment-emergent adverse events (TEAEs) were cytokine release syndrome (CRS; all Grade 1: 44%) and neutropenia (any Grade: 38%; Grade 3/4: 33%). Among other adverse events of special interest, one patient in the 50 mg cohort experienced Grade 1 immune effector cell-associated neurotoxicity syndrome (ICANS). Infections occurred in 84% of patients (Grade 1/2: 51%; Grade 3: 33%) with the majority occurring within the first three months of treatment and the rate of infections decreased over time. There were no ≥Grade 4 infections, Grade 5 TEAEs or dose-limiting toxicities. Ten patients elected to undergo an autologous stem cell transplant, all of whom had an acceptable CD34+ stem cell yield post-induction (range: 2.5-11.5 x 106/kg).
A broad clinical development program investigating Lynozyfic in early stages of the disease is underway. This includes the Phase 2 portion of the LINKER-MM4 trial evaluating Lynozyfic at the recommended 200 mg dose, as well as LINKER-MM6 (EMN39), a trial evaluating a combination of daratumumab, lenalidomide and dexamethasone (DRd) followed by Lynozyfic monotherapy compared with continued DRd in transplant-ineligible NDMM.
The use of Lynozyfic described above is investigational, and its safety and efficacy has not been evaluated by any regulatory authority for this indication.
About the ‘Regeneron Roundtable’ Investor Event
Links to the webcast and to register via telephone may be accessed from the ‘Investors and Media’ page of Regeneron’s website at https://investor.regeneron.com/events-and-presentations. Upon registration, all telephone participants will receive a confirmation email detailing how to join the conference call, including the dial-in number along with a unique passcode and registrant ID that can be used to access the call. A replay of the conference call and webcast will be archived on the company’s website for at least 30 days.
About Multiple Myeloma
As the second most common blood cancer, there are over 187,000 new cases of MM diagnosed globally every year, with more than 36,000 diagnosed and 12,000 deaths anticipated in the U.S. in 2025. The disease is characterized by the proliferation of cancerous plasma cells (MM cells) that crowd out healthy blood cells in the bone marrow, infiltrate other tissues and cause potentially life-threatening organ injury. Despite treatment advances, MM is not curable, and while current treatments are able to slow progression of the cancer, most patients will ultimately experience cancer progression and require additional therapies.
About Lynozyfic
Lynozyfic was invented using Regeneron’s VelocImmune® technology and is a fully human BCMAxCD3 bispecific antibody designed to bridge B-cell maturation antigen (BCMA) on MM cells with CD3-expressing T cells to facilitate T-cell activation and cancer-cell killing. Lynozyfic is approved to treat certain adults with R/R MM: in the
In the U.S., the generic name for Lynozyfic in its approved indications is linvoseltamab-gcpt, with gcpt as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. FDA. Outside of the U.S., the generic name of Lynozyfic in its approved indications is linvoseltamab.
Lynozyfic is being investigated in a broad clinical development program exploring its use as a monotherapy as well as in combination regimens across different lines of therapy in MM, including earlier lines of treatment, as well as plasma cell precursor disorders. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
In addition to LINKER-MM4, ongoing trials include:
For more information on Regeneron’s clinical trials in blood cancer, visit the clinical trials website, or contact via clinicaltrials@regeneron.com or 844-734-6643.
IMPORTANT SAFETY INFORMATION FOR
What is the most important information I should know about LYNOZYFIC?
LYNOZYFIC may cause serious or life-threatening side effects, including Cytokine Release Syndrome (CRS) and infusion-related reactions (IRR), or neurologic problems.
Cytokine Release Syndrome (CRS) and infusion related reactions (IRR). CRS is common during treatment with LYNOZYFIC and can also be serious or life-threatening. Tell your healthcare provider or get medical help right away if you develop any signs or symptoms of CRS or IRR, including:
|
|
|
|
Neurologic problems. LYNOZYFIC can cause neurologic problems that can be serious or life-threatening. Tell your healthcare provider or get medical help right away if you develop any signs or symptoms of neurologic problems, including:
Due to the risk of CRS and neurologic problems, you will receive LYNOZYFIC on a “step-up dosing schedule” and should be hospitalized for 24 hours after the first and second “step-up” doses.
LYNOZYFIC is available only through the LYNOZYFIC Risk Evaluation and Mitigation Strategy (REMS) due to the risk of side effects of CRS and neurologic problems. You will receive a
Your healthcare provider will monitor you for signs and symptoms of CRS and neurologic problems during treatment with LYNOZYFIC, as well as other side effects, and may treat you in a hospital if needed. Your healthcare provider may temporarily stop or completely stop your treatment with LYNOZYFIC if you develop CRS, neurologic problems, or any other severe side effects.
If you have any questions about LYNOZYFIC, ask your healthcare provider.
Before receiving LYNOZYFIC, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive LYNOZYFIC?
What should I avoid while receiving LYNOZYFIC?
Do not drive, or operate heavy or potentially dangerous machinery, or do other dangerous activities for 48 hours after completing each of your “step-up” doses or at any time during treatment with LYNOZYFIC if you develop new neurologic symptoms, until the symptoms go away.
What are the possible side effects of LYNOZYFIC?
LYNOZYFIC may cause serious side effects, including:
The most common side effects of LYNOZYFIC include:
|
The most common severe abnormal blood test results with LYNOZYFIC include: low white blood cell counts and low red blood cell counts.
These are not all of the possible side effects of LYNOZYFIC.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
Please see full Prescribing Information, including Boxed WARNING, and Medication Guide for LYNOZYFIC.
What is LYNOZYFIC?
LYNOZYFIC is a prescription medicine used to treat adults with multiple myeloma who:
It is not known if LYNOZYFIC is safe and effective in children.
About
At Regeneron, we’re applying more than three decades of biology expertise with our proprietary VelociSuite® technologies to develop medicines for patients with diverse blood cancers and rare blood disorders.
Our blood cancer research is focused on bispecific antibodies that are being investigated both as monotherapies and in various combinations and emerging therapeutic modalities. Together, they provide us with unique combinatorial flexibility to develop customized and potentially synergistic cancer treatments.
Our research and collaborations to develop potential treatments for rare blood disorders include explorations in antibody medicine, gene editing and gene-knockout technologies, and investigational RNA-approaches focused on depleting abnormal proteins or blocking disease-causing cellular signaling.
About
About
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to numerous approved treatments and product candidates in development, most of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, neurological diseases, hematologic conditions, infectious diseases, and rare diseases.
Regeneron pushes the boundaries of scientific discovery and accelerates drug development using our proprietary technologies, such as VelociSuite, which produces optimized fully human antibodies and new classes of bispecific antibodies. We are shaping the next frontier of medicine with data-powered insights from the Regeneron Genetics Center® and pioneering genetic medicine platforms, enabling us to identify innovative targets and complementary approaches to potentially treat or cure diseases.
For more information, please visit www.Regeneron.com or follow
Forward-Looking Statements and Use of Digital Media
This press release includes forward-looking statements that involve risks and uncertainties relating to future events and the future performance of Regeneron Pharmaceuticals, Inc. (“Regeneron” or the “Company”), and actual events or results may differ materially from these forward-looking statements. Words such as “anticipate,” “expect,” “intend,” “plan,” “believe,” “seek,” “estimate,” variations of such words, and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. These statements concern, and these risks and uncertainties include, among others, the nature, timing, and possible success and therapeutic applications of products marketed or otherwise commercialized by

![]()
Source: Regeneron Pharmaceuticals, Inc.
Treatment with INCA033989 as monotherapy or in combination with ruxolitinib (Jakafi) was well tolerated and led to spleen and anemia responses in patients with CALR exon 9–mutated myelofibrosis who were resistant or intolerant to prior JAK inhibitor therapy, or were ineligible for JAK inhibitor therapy, according to data from the phase 1 INCA033989-101 (NCT05936359) and INCA033989-102 (NCT06034002) trials.1
Findings presented at the
In patients treated with INCA033989 plus ruxolitinib (n = 20), all had any-grade TEAEs, including 65.0% with any-grade TRAEs, 55.0% with grade 3 or higher TEAEs, and 25.0% with serious TEAEs. No DLTs were reported with the combination. TEAEs led to treatment discontinuation in 10.0% of patients, dose reductions in 5.0% of patients, infusion interruption in 5.0% of patients, and dose delays in 40.0% of patients.
Regarding efficacy, INCA033989 monotherapy (n = 36) yielded a spleen volume reduction of at least 25% (SVR25) at week 24 in 41.7% of patients and an SVR of at least 35% (SVR35) at week 24 in 33.3% of patients. Best SVR was 47.9% for SVR25 and 31.3% for SVR35. In patients without any prior JAK inhibitor exposure (n = 7), the 24-week SVR25 and SVR35 rates were 71.4% and 57.1%, respectively. In patients who were relapsed/refractory or intolerant to a JAK inhibitor (n = 29), these respective rates were 34.5% and 27.6%.
For patients treated with the combination, evaluable patients at week 24 (n = 12) experienced SVR25 and SVR35 rates of 50% and 25%, respectively. A best SVR of SVR25 occurred in 11 total patients; SVR35 was reported in 8 patients.
“INCA033989 is really well tolerated both as monotherapy and in combination with ruxolitinib,” lead study author John O. Mascarenhas, MD, said in a presentation of the data. “We saw rapid spleen and anemia responses in both cohorts.”
Mascarenhas is a professor of medicine at the Icahn School of Medicine at Mount Sinai in New York, New York, where he also serves as director of the Center of Excellence for Blood Cancers and Myeloid Disorders. He is also a member of The Tisch Cancer Institute, where he is director of the Adult Leukemia Program and leader of clinical investigation in the Myeloproliferative Disorders Program.
Mascarenhas reported that approximately 25% to 35% of patients with myelofibrosis harbor CALR exon 9 mutations, and higher CALR variant allele frequency (VAF) has been associated with more advanced disease featuring anemia and elevated peripheral blasts.
INCA033989—a novel, fully human, high-affinity, Fc-silenced, immunoglobulin G1 monoclonal antibody—is a therapy designed to target CALR exon 9 mutations through selective targeting in complex with the thrombopoietin receptor.
INCA033989-101 is being conducted outside the United States (US), while INCA033989-102 is including US patients specifically. Both studies are enrolling patients at least 18 years of age with primary or post–essential thrombocythemia myelofibrosis harboring CALR exon 9 mutations.1-3
Patients need to have a spleen volume of at least 450 mL or palpable splenomegaly of at least 5 cm.1 To be included in the monotherapy arm, patients need to be intolerant to a JAK inhibitor, resistant to a JAK inhibitor for at least 12 weeks, or ineligible for a JAK inhibitor. Those in the combination arm needed to have a suboptimal response to ruxolitinib given for at least 12 weeks of prior treatment.
During dose escalation, patients in both arms received INCA033989 at 24 mg to 2500 mg intravenously once every 2 weeks. The dose-expansion portion of the study includes monotherapy and combination arms, along with a randomized portion where JAK inhibitor–naive patients are being randomly assigned to INCA033989 with or without ruxolitinib.
The primary end points of dose escalation are evaluating the incidence of DLTs and TEAEs. Secondary end points comprised SVR25 and SVR35 at weeks 12 and 24; anemia response; symptom improvement; and change in CALR exon 9 VAF.
In the monotherapy and combination arms, the median age was 59.5 years (range, 34-76) and 61.0 years (range, 38-82), respectively. The rates of female patients were 32.7% and 20.0%, respectively, and patients had respective median times from initial diagnosis of 7.4 years (range, 0-25.3) and 3.1 years (range, 0.4-16.4). CALR exon 9 mutation type included type 1 (monotherapy arm, 57.7%; combination arm, 60.0%), type 2 (21.2%; 35.0%), and other (21.2%; 5.0%). The median CALR VAF was 36% (range, 24%-53%) in the monotherapy arm and 39% (range, 30%-85%) in the combination arm.
In the monotherapy arm, 93.3% of evaluable patients (n = 45) experienced symptom improvements, and 60.0% achieved at least a 50% reduction in total symptom score (TSS50).
For those evaluable for anemia response (n = 25), best responses comprised major response (40.0%), minor response (16.0%), stable anemia (32.0%), progressive anemia (8.0%), and missing (4.0%). In patients with transfusion-dependent anemia, best responses were major response (20.0%), minor response (40.0%), stable anemia (20.0%), and progressive anemia (20.0%). Those without transfusion-dependent anemia (n = 20) had best anemia responses of major response (45.0%), minor response (10.0%), stable anemia (35.0%), progressive anemia (5.0%), and missing (5.0%).
Notably, a reduction of CALR exon 9 mutation VAF occurred in 89.4% of evaluable patients in the monotherapy arm (n = 47), and 10.6% of patients achieved a best reduction of at least 25%.
In the combination arm, 81.3% of patients (n = 16) experienced symptom improvements, and 33.3% (n = 9) achieved TSS50 at week 24. In evaluable patients (n = 14), 86% had stable anemia during the study, and 1 patient had a major anemia response.
In the INCA033989 monotherapy group, the most common any-grade TEAEs reported in at least 15% of patients included anemia (30.8%), fatigue (26.9%), thrombocytopenia (25.0%), arthralgia (21.2%), increased aspartate aminotransferase (AST; 21.2%), cough (21.2%), diarrhea (21.2%), headache (21.2%), leukopenia (21.2%), nausea (21.2%), pruritus (21.2%), hyperglycemia (19.2%), neutropenia (19.2%), nasal congestion (15.4%), and extremity pain (15.4%).
In the combination arm, the most frequent TEAEs of any grade that occurred in at least 15% of patients consisted of anemia (45.0%), thrombocytopenia (35.0%), increased alanine aminotransferase levels (20.0%), diarrhea (20.0%), fatigue (20.0%), increased AST (15.0%), and cough (15.0%).

Rawalpindi [Pakistan], December 8 (ANI): Local public transporters in Rawalpindi and Islamabad have announced a wheel-jam strike today, rejecting the administration’s request to postpone their protest against a new traffic ordinance that…
