- US Department of Labor applauds President Trump’s bold directive to rein in politicization of capital markets by proxy services U.S. Department of Labor (.gov)
- Trump gives Elon Musk a win over a longtime foe CNN
- Proxy Advisors in the Crosshairs. Securities Law Implications of New Executive Order The National Law Review
- GC Cheat Sheet: The Hottest Corporate News Of The Week Law360
- White House’s Executive Order on Proxy Advisors: 7 Things to Know Now JD Supra
Category: 3. Business
-
US Department of Labor applauds President Trump’s bold directive to rein in politicization of capital markets by proxy services – U.S. Department of Labor (.gov)
-
SPIE announces the success of its SHARE FOR YOU 2025 employee shareholding plan
Cergy, 12th December 2025 – SPIE, the independent European leader in multi-technical services in the areas of energy and communications, announces the success of its SHARE FOR YOU 2025 employee shareholding plan.
SPIE also announces its intention to implement an anti-dilutive share buyback program
Employee shareholding is part of SPIE’s culture and this year’s new plan has been a remarkable success. Employees’ participation increased significantly: close to 25,000 employees from 17 countries subscribed to the offer (versus around 21,000 employees in 2024). More than 6,000 employees invested for the first time, including people stemming from recently acquired companies.
The employee contributions to the 2025 SHARE FOR YOU plan amounted to 62 million euros. Following this operation completed, on 12 December 2025, 2,101,883 new shares have been issued by the company. For this new SHARE FOR YOU plan, which ran from 25 September to 16 October 2025, SPIE’s employees benefitted from a 20% discount[1].
After the SHARE FOR YOU 2025 plan, more than one employee in two is a Group shareholder and the share of capital held by employees thanks to these programs is approximately 8%.
Gauthier Louette, Chairman and CEO of SPIE, declared:
“Through their strong engagement in the 2025 employee shareholding program, our teams reaffirm their trust and commitment to SPIE. The Executive Committee and I extend our sincere gratitude.
We take great pride in this dynamic. Our people are the cornerstone of our success, and this initiative reflects the entrepreneurial spirit that drives our Group forward. Together, we are shaping an ambitious and sustainable future.”
While maintaining a strong discipline regarding its leverage ratio, the Group plans to implement early in 2026 a share buyback program which will partially compensate the dilutive impacts implied by the employee shareholding program and the long-term incentive plan. Details of this share buyback program will be provided further in a dedicated press release.
[1] Subscription price at €38.55 after discount
Continue Reading
-

WTAS: Financial Services Highlights Support for Committee’s Bipartisan INVEST Act – U.S. House Financial Services Committee (.gov)
- WTAS: Financial Services Highlights Support for Committee’s Bipartisan INVEST Act U.S. House Financial Services Committee (.gov)
- House passes INVEST Act to ease investment standards and boost capital in markets CNBC
- Bloomberg Talks: Rep. Gregory Meeks Bloomberg.com
- RELEASE: HILL VOTES TO EMPOWER ARKANSAS FAMILIES AND SMALL BUSINESSES Congressman French Hill (.gov)
- House bill would compel SEC to revise small RIA regulation Citywire
Continue Reading
-

ULM computer science students win Nexus Louisiana’s DevDays HealthTech Challenge
Published December 12, 2025
CAPTION: Champions of the DevDays HealthTech Challenge, Yukta Karki, SianRose Vincent, and Nirjara KC, pictured with their faculty advisor, Prasanthi Sreekumari, awarded $5,000 prize.
MONROE, La. – The ULM Department of Computer Science is proud to announce that a student team has
won the DevDays HealthTech Challenge, hosted by Nexus Louisiana in partnership with
Ochsner Health. Kneva, the winning team composed of computer science students Yukta
Karki, SianRose Vincent, and Nirjara KC, was awarded a $5,000 prize at the competition
in Baton Rouge on November 14.The DevDaysChallenge attracted 160 students from 11 Louisiana universities who submitted 45
innovative solutions, addressing issues ranging from athlete safety to carbon management.Kneva is an innovative health technology platform focused on addressing a long-overlooked
crisis in sports medicine: female athletes face up to eight times greater risk of
experiencing a catastrophic ACL injury. This groundbreaking project aims to end the
era of compromise in women’s sports by delivering technology and insights that support
safer performance and long-term athletic health.The system unifies two critical data points, hormonal fluctuations and biomechanical
movement, to predict elevated injury risk and deliver real-time, micro-adjusted training
feedback. A wearable knee sensor monitors movement patterns, while the platform maps
an athlete’s menstrual cycle to identify when they are more susceptible to injury,
creating a personalized, adaptive approach to performance and injury prevention.Team Kneva shared that the competition affirmed their mission to build injury-prevention
tools that truly reflect the physiology and experiences of female athletes—not systems
retrofitted from male data.“I was raised in a world shaped by strong women, and Kneva lets me pour that energy
into a platform that finally acknowledges and supports female athletes,” said KC.
“Leading the research and development for our ACL prevention prototype proved that
innovation is iterative, and this win fuels our next chapter. We’re excited to take
on the engineering challenges needed to move from concept to a production-ready solution.”The six-week competition brought together student teams from across the state to confront
a real and pressing sports medicine issue. “DevDays is about putting our state’s brightest
minds on our state’s hardest problems,” said Tony Zanders, President and CEO of Nexus
Louisiana. “The quality of ideas we saw reflects the strength of the talent emerging
from Louisiana’s universities and the value of connecting that talent to real industry
needs.”As the presenting partner, Ochsner Health emphasized the importance of strengthening
pathways between students and industry. “DevDays shows what happens when we give young
people real problems to solve and the room to innovate boldly. These students aren’t just
imagining the future of sports medicine — they’re building it. I’m proud that Ochsner
is helping open doors, strengthen pathways, and champion the next generation of Louisiana
talent,” said Christy Reeves, Vice President, Network Development and Government Relations
at Ochsner Health.“We are extremely proud of all the students who participated in the competition. Their
dedication, passion, and perseverance showcase the strength and spirit of our department,”
said Dr. Prasanthi Sreekumari, Program Chair of the ULM Computer Science Department
and Associate Professor of Computer Science.“Kneva’s victory is a powerful validation of their mission and the impact their work
can have on the future of sports medicine,” added Sreekumari.“The team would like to thank Dr. Sreekumari for her guidance throughout the development
of Kneva. We also extend our thanks to Dr. Paul Wiedemeier and Dr. Paul Rojas for
providing the resources and support we needed to bring this project to life,” said
KC.Team Kneva’s win marks the second victory for a ULM Computer Science team at DevDays this
year. Carbon Horizon won the Nexus DevDays ClimateTech Challenge in October 2025.
Read more about their win at https://www.ulm.edu/news/2025/comp_science_devdays_111125.htmlAbout Nexus Louisiana DevDays
Nexus Louisiana’s mission is to accelerate the growth of high-potential technology-enabled
companies by providing them with coaching, capital, and connections. Their programs leverage regional
collaboration to foster mentorship, investment, and opportunity for local entrepreneurs
to grow their ideas and transform the technology industry in Louisiana.DevDays are one-of-a-kind events that fuse technology, competition, and Louisiana’s
legendary football pride. Over six weeks, participants tackle real-world challenges
from leading industries and bring their solutions to life in an impactful, one-day
demo event in Baton Rouge.Nexus Louisiana has confirmed that DevDays will return next year with new challenges
and expanded collaboration opportunities.For more information, please visit https://nexusla.org/devdays.
Continue Reading
-

Inside Edelman: New Places, New Perspectives – Megan Johnson
From Canada to the U.S., India, and now Singapore — Megan’s global journey reflects a deep curiosity, a love of change, and an openness to the unexpected. Each move has broadened her perspective, strengthened her adaptability, and deepened her appreciation for the shared Edelman spirit that connects teams across continents. As she settles into life in Singapore, she reflects on the experiences, relationships, and lessons that continue to shape both her career and her understanding of the world.
What is something you’ve discovered about yourself during your time abroad?
As part of my Edelman career, I’ve had the wonderful opportunity of working in different offices across different countries – from Canada to the US, India, and now Singapore. While I wouldn’t say I “discovered” this necessarily, but my travels have certainly reinforced how much I like change. Trying something new, embracing the unexpected, and being uncomfortable all help open your eyes to what the world has to offer.
How has the experience of working in a new market expanded your understanding of Edelman’s global work or approach?
What’s struck me in every office I’ve worked in is the balance between familiarity and local uniqueness. Each office feels connected through our shared values, yet every local market brings its own voice and way of doing things. Experiencing those differences has shown me how diverse perspectives strengthen our global work and make our network richer and more dynamic.
Is there a favorite local custom, place, or routine that has become meaningful to you in your host country?
I’m still getting settled in Singapore, but if I look at my time in India … that experience will always hold tremendous meaning for me – my first leap into an adventure very far away from home. The sights, the smells, the sounds (I will forever be desensitized to honking), the food (!), the laughs, and warmth of all of those who helped me along the way – I couldn’t be more thankful.
What new skills, perspectives, or ways of working have you gained from your host team?
Understanding how to work with different cultures is fascinating. No country is alike, but the diversity of Asia in particular keeps you on your toes. You have to listen first, be patient, adaptable, open to changing how you might typically do things, and roll with it. And most importantly – have a good laugh as you learn (and make a few embarrassing mistakes) along the way.
What is one highlight or proud moment from your time abroad that you’ll carry forward in your career?
It comes down to the relationships. I have met, worked with, been in the trenches with, and become lifelong friends with so many colleagues over the years. Those partnerships, experiences, and memories are everlasting, and have – and will continue to shape – all aspects of my life and career.
Megan Johnson is a Global Client Leader for APAC based in the Singapore office.
Continue Reading
-

Authorities launch joint plan to get more people working
A NEW 10-year plan has been launched to help thousands more people across Leicester, Leicestershire and Rutland (LLR) find good jobs, earn more money, and live healthier, happier lives.
The Get Leicester, Leicestershire & Rutland Working Plan 2025-2035 aims to improve economic opportunities and tackle the barriers that prevent residents from getting good quality work.
It’s been led by Leicester City Council, working in partnership with Leicestershire County Council, Rutland County Council, the Department for Work and Pensions (DWP), and the NHS Integrated Care Boards (ICB) for Leicester, Leicestershire & Rutland.
The authorities have also worked alongside local councils, employers, colleges and community organisations to ensure the plan is focused on helping everyone to benefit from a stronger, fairer local economy.
The plan sets out four key priorities for action:
Partnership working: Continue to build a strong partnership working between councils, the NHS, employers and the VCSE sector to deliver a seamless, coordinated employment, health and skills system.
Evidence-led delivery: Use shared data, intelligence, insight and evaluation to target resources effectively and scale place-based approaches that deliver measure impact.
Employer engagement: Engage businesses proactively to align employment support and provision with labour market needs, promote inclusion and drive economic growth.
Breaking down barriers: Continue to help to break down economic inactivity barriers linking to growth opportunities and creating clear, supported pathways to secure, high-quality employment.
Cllr Elly Cutkelvin, Leicester deputy city mayor for housing, economy and neighbourhoods, said: “The Government has set us the challenging target of raising employment levels to 80%, but it is a challenge we are embracing.
“Through this plan, we are making a commitment to the people of the region that if they step up, we will work across public services to support them, in whatever ways they need. It’s why we are working so closely with our partners in health, skills, employment support, and in the benefits system.
“We realise this is a challenging time for the economy across the area, but we will continue to work together to maximise the opportunities and resources across LLR to support our residents into employment opportunities.”
Deputy leader of Leicestershire County Council, Kevin Crook, said: “We’re pleased to be supporting this partnership plan, which is bringing together a number of organisations to spot and create as many opportunities to help people into employment.
“We know that across the county there is huge potential, and we know that businesses know their needs. That’s why this evidence-based plan will build on our strengths by bringing together partners and business leaders to create sustainable employment opportunities, fill vacancies and support economic growth.”
Cllr Rosemary Powell, portfolio holder for property and economic development at Rutland County Council, said: “Building a strong rural economy with a productive, sustainable and diverse business base is a key pillar of our long-term strategy for a thriving county. To achieve this, it’s important we understand the needs of local businesses and make sure training, education and funding opportunities are geared towards meeting these needs. The Get Leicester, Leicestershire & Rutland Working Plan will play a really important role in helping us to grow a more productive local economy – working alongside our business partners to provide greater opportunities for better paid jobs locally.”
Laura Moig, group director for DWP Central Midlands said: “This working plan was developed by a strong partnership and is our strategy to reduce economic inactivity and increase employment across LLR. The plan has a real focus on improving health, work and skills to create more opportunities for everyone.”
Professor Nil Sanganee, chief medical officer of the Leicester, Leicestershire and Rutland Integrated Care Board, said: “People experiencing ill health are a key priority for this plan, including those with long-term health conditions or disabilities and those undergoing planned or unplanned episodes of care. Supporting these individuals to enter, remain in, or return to work is essential, both for improving population health outcomes and for strengthening the economic resilience of our communities.
“NHS partners in LLR have already demonstrated a strong commitment to addressing the vital links between work and health, through initiatives such as Work Well, which aims to support people with long-term conditions to enter or remain in employment.
“We will work collaboratively with local authorities, employers and system partners to ensure that health is never a barrier to opportunity. Together, we can create the conditions where everyone has the opportunity to work, contribute and live well.”
The plan is a live document that will grow and adapt as the economy and local priorities evolve. Regular reviews will ensure the plan stays relevant, flexible, and focused on making a lasting difference.
The full plan is available on the city council’s website at leicester.gov.uk/get-llr-working
Continue Reading
-
Exclusive: FDA leaders pushed to cut Lilly weight-loss pill review time – Reuters
- Exclusive: FDA leaders pushed to cut Lilly weight-loss pill review time Reuters
- U.S. Regulators Pushed to Fast-Track Approval of Eli Lilly’s Weight-Loss Pill: Reports TipRanks
- FDA reportedly pressing for reviewers to speed up evaluation of Eli Lilly’s weight-loss pill StreetInsider
- FDA May Fast-Track Eli Lilly’s Weight Loss Pill – Eli Lilly (NYSE:LLY) Benzinga
- Exclusive-US FDA brass pushed internally for speedier Lilly weight-loss pill verdict 104.1 WIKY
Continue Reading
-

Pak, Binance sign pact for ‘tokenisation’ of assets up to USD 2 billion – Press Trust of India
- Pak, Binance sign pact for ‘tokenisation’ of assets up to USD 2 billion Press Trust of India
- Pakistan to allow Binance to explore ‘tokenisation’ of up to $2bn of assets Dawn
- Binance and Pakistan Partner to Advance Digital-Asset Innovation and Regulatory Development Binance
- Bilal Bin Saqib: The Youngest Technocrat Driving Pakistan’s Leap Into the Digital Economy FF News | Fintech Finance
- NOCR 2025: a critical review Business Recorder
Continue Reading
-
A New Era of Care: How AI Is Shaping the Patient and Clinician Experience – Epic Systems
- A New Era of Care: How AI Is Shaping the Patient and Clinician Experience Epic Systems
- Cut Through The Hype: 3 Things To Know Before Adopting Agentic AI MedCity News
- Artificial Intelligence and Healthcare La Voz de Lanzarote
- AI Evening: AI in Healthcare: Building Trust & Driving Real-World Impact Coastal View News
- Will AI become medicine’s new operating system? Digital Journal
Continue Reading
-
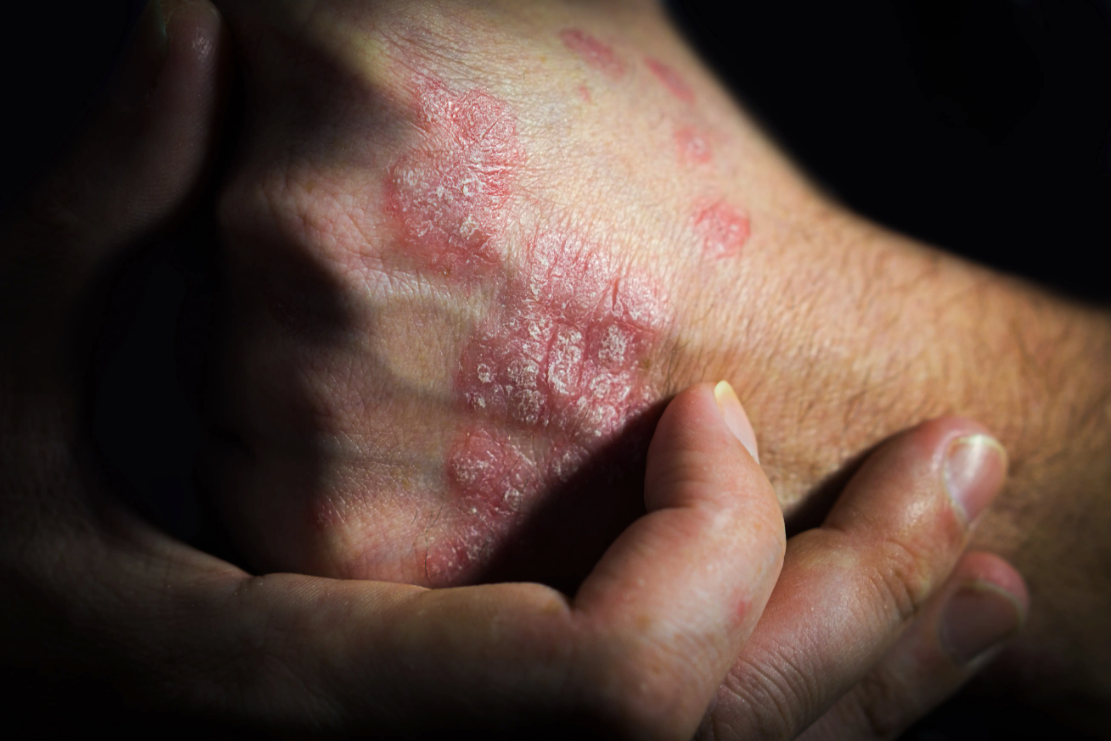
Real-World Safety Signals of Bimekizumab Emerge in New FAERS Analysis
Bimekizumab, a dual IL-17A/IL-17F inhibitor used for
psoriasis and psoriatic arthritis, is increasingly used in real-world practice, prompting closer evaluation of its long-term safety.1 In an analysis from the FDA Adverse Event Reporting System (FAERS) covering the third quarter (Q3) of 2021 to the fourth quarter (Q4) of 2024, researchers identified 2780 bimekizumab-related reports and 70 significant safety signals, including both known and newly detected adverse events (AEs).This retrospective analysis is published in
Naunyn-Schmiedeberg’s Archives of Pharmacology .“Given the limited time since its market approval, there is a scarcity of long-term safety data based on real-world studies,” wrote the researchers of the study. “To address this gap, we utilized the FAERS database to identify and analyze bimekizumab-related AEs, thereby enhancing our understanding of its safety profile in routine clinical practice.”
In the US, bimekizumab was first authorized in October 2023 for adults with moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy, based on phase 3 trial data demonstrating high levels of skin clearance.2 Since then, US approvals have expanded to include active psoriatic arthritis, active nonradiographic axial spondyloarthritis with objective signs of inflammation, and active ankylosing spondylitis.3 These indications position bimekizumab as one of the first dual IL-17A/IL-17F inhibitors available for a range of immune-mediated inflammatory disorders.
This study used a retrospective pharmacovigilance design to evaluate AEs associated with bimekizumab using data from the FAERS.1 All reports submitted between Q3 2021 and Q4 2024 were extracted and screened for cases listing bimekizumab as the primary suspect drug. Analyses were conducted to identify statistically significant safety signals across system organ classes, using established metrics to compare the frequency of reported events with those of the full FAERS database. Time-to-onset analyses were also performed to characterize when adverse events occurred following drug initiation.
Among the 6,037,398 AE reports submitted to FAERS during the study period, 2780 were associated with bimekizumab. The researchers identified 70 significant adverse event signals across 11 System Organ Classes. Many aligned with the drug’s known safety profile, including injection site pain, oral candidiasis, and esophageal candidiasis. However, 29 previously unreported signals also emerged, such as cellulitis, lower respiratory tract infections, Staphylococcus infections, immunodeficiency, and Mycoplasma pneumonia.
Time-to-onset analysis showed a median (IQR) onset of 29 (0, 84.75) days, indicating that many events occurred within the first several weeks of therapy.
However, the researchers acknowledged several limitations. First, FAERS reports are often incomplete, leading to missing data that may bias both AE patterns and time-to-onset findings. Because information on patients taking bimekizumab without AEs was unavailable, true incidence rates could not be calculated. Additionally, the presence of confounders, such as concomitant drugs or underlying conditions, further limited interpretability of this retrospective study, so controlled clinical studies are needed to confirm these safety signals.
Despite these findings, the researchers believe these findings confirm established risks while revealing additional safety signals that warrant further clinical attention in bimekizumab.
References
1. Lin X, Guo L, Lin T, et al. Disproportionality analysis of adverse events associated with bimekizumab: a real-world study based on FDA Adverse Event Reporting System (FAERS) Database. Naunyn Schmiedebergs Arch Pharmacol. doi:10.1007/s00210-025-04894-2
2. Myshko D. Bimekizumab-bkzx, the newest psoriasis treatment, is now available. AJMC®. November 30, 2023. Accessed December 11, 2025.
https://www.ajmc.com/view/bimekizumab-bkzx-the-newest-psoriasis-treatment-is-now-available 3. McNulty R. FDA approves bimekizumab for psoriatic arthritis, nonradiographic axSpA, ankylosing spondylitis. AJMC. September 23, 2024. Accessed December 11, 2025.
https://www.ajmc.com/view/fda-approves-bimekizumab-for-psoriatic-arthritis-non-radiographic-axspa-ankylosing-spondylitis Continue Reading
Tweet