Background
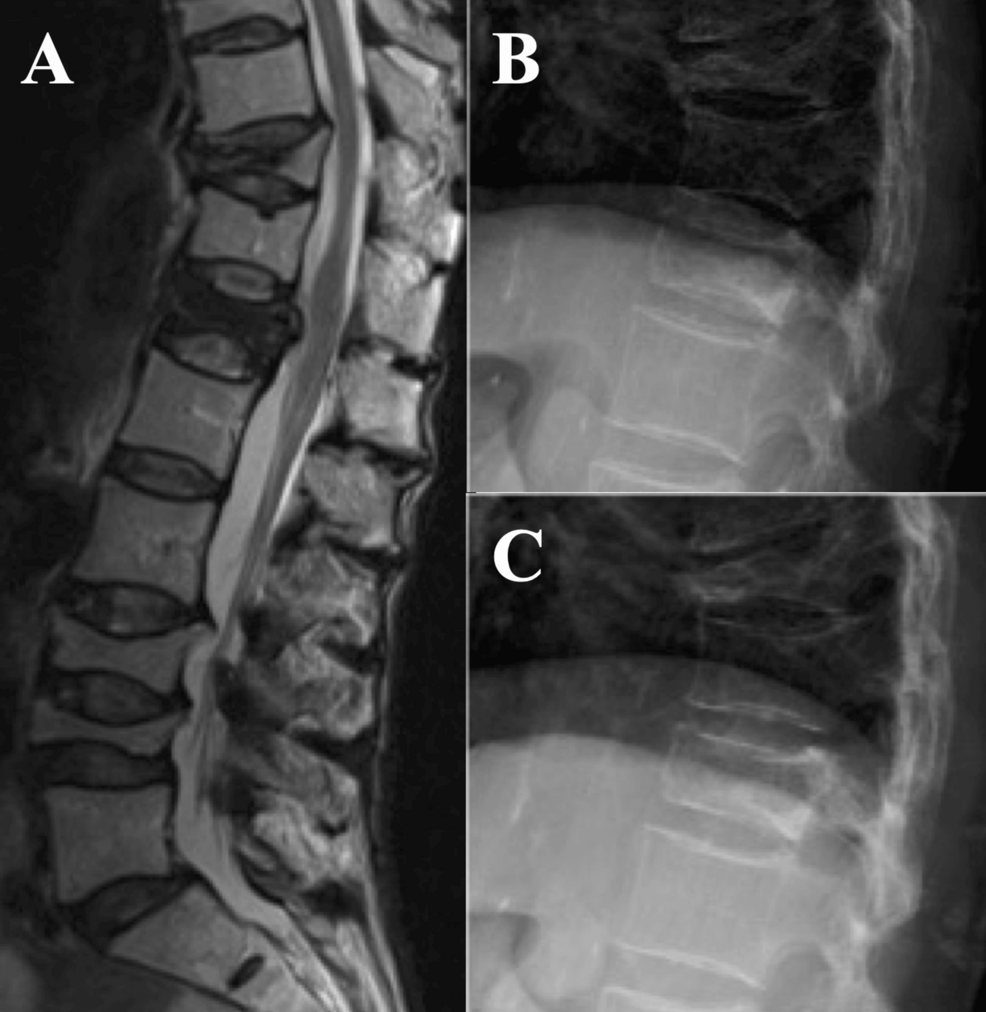
Colorectal cancer (CRC) is the third most common malignancy worldwide, with over 1.9 million new cases and 935,000 deaths annually.1 Accurate staging using the Tumor-Node-Metastasis (TNM) system is critical for prognostic…
Colorectal cancer (CRC) is the third most common malignancy worldwide, with over 1.9 million new cases and 935,000 deaths annually.1 Accurate staging using the Tumor-Node-Metastasis (TNM) system is critical for prognostic…
Chronic prostatitis/chronic pelvic pain syndrome (CP/CPPS) is characterized by urogenital pain or discomfort lasting at least 3 months,1 and is a type of genitourinary pain caused by non-bacterial infections with predominantly…

Children exposed to high levels of screen time before age two showed changes in brain development that were linked to slower decision-making and increased anxiety by their teenage years, according to new research by Asst Prof Tan Ai…
Bullous pemphigoid is a chronic, subepidermal autoimmune blistering disorder, the pathogenesis involves autoantibodies targeting hemidesmosomal proteins BP180 and BP230, which are essential for dermal–epidermal adhesion.1 Patients…
Ramsay Hunt Syndrome is a neurological condition caused by the reactivation of the varicella-zoster virus (VZV) in the facial nerve’s geniculate ganglion (cranial nerve VII).1 Vesicular eruptions in the mouth or ear, ear pain, and…

KUALA LUMPUR, Dec 31 — Male infertility is still treated as a taboo in many Malaysian households, but clinicians at Hospital Canselor Tuanku Muhriz (HCTM) UKM say more men have begun seeking assessment in…
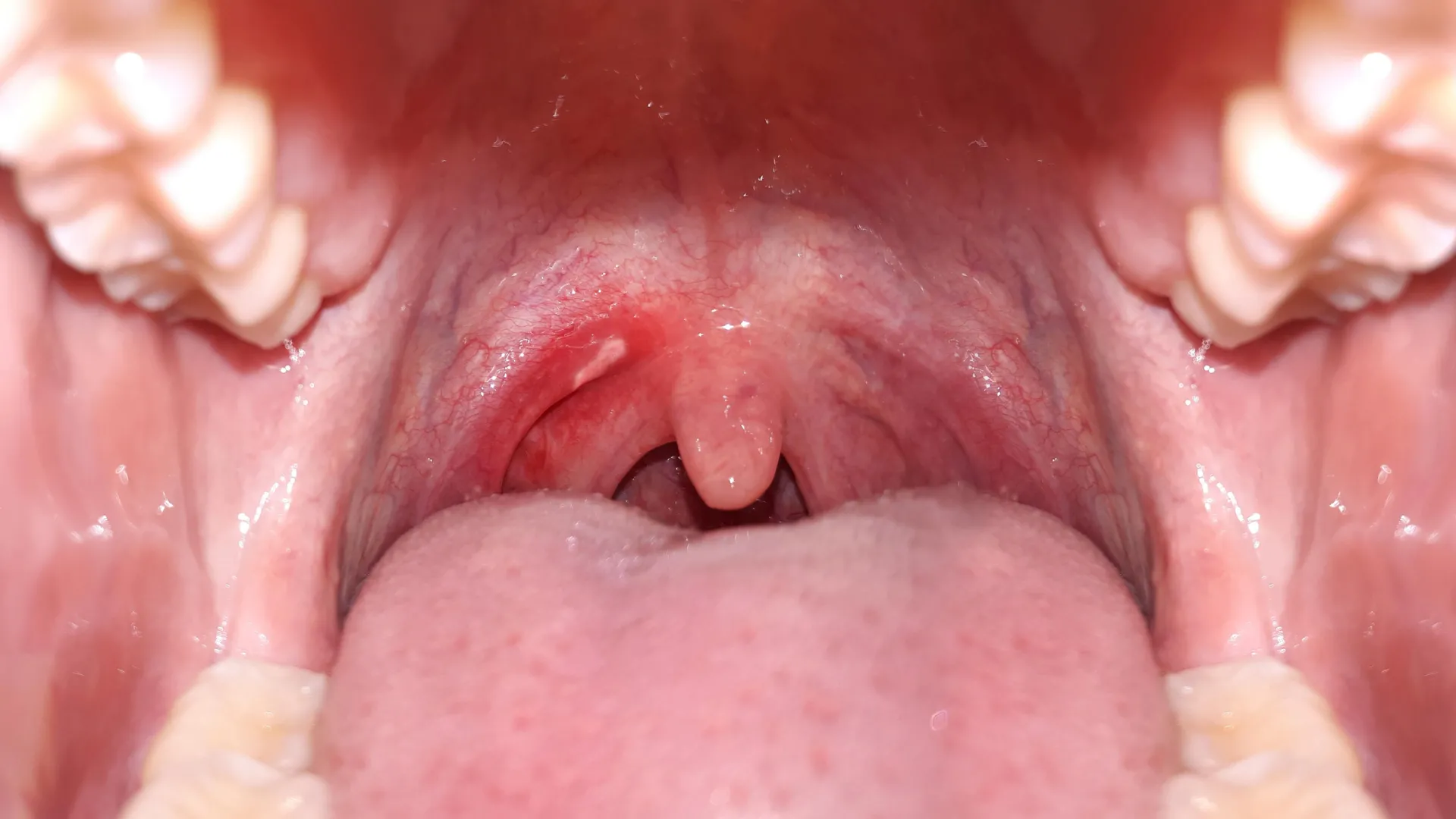
A large comparative study published online in the open access journal BMJ Global Health has found that even low daily alcohol consumption is linked to a much higher risk of mouth cancer in India. Drinking just 9 g of alcohol per day, about the…

The fungal species Candida auris is spreading across the globe, and gaining in virulence, according to a new review by a Hackensack Meridian Center for Discovery and Innovation (CDI) scientist and colleagues.
But there are…

NYT The virus, sometimes called the stomach bug, is incredibly contagious. Here’s how to stay safe this season. Every year, there’s a surge in norovirus cases between November and April, and this holiday season is no…