Mice
C57BL/6 mice, purchased from SPF (Beijing) Biotechnology Co., were used in all experiments. Mice were group-housed (four per cage) and given free access to water and food under controlled environmental conditions at 24 ± 2 °C with 50% relative humidity, and on a 12 h-light/dark cycle with light on from 08:00 to 20:00. All experimental procedures involving the use of animals were approved by the Ethics Committee on Animal Experiments at Jinan University. All animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals. No formal randomization was used, and no animals were excluded. For drug treatment, mice were first tested for social interaction and weighed. Animals were then stratified based on sociability and body weight, and randomly assigned to drug or vehicle groups within each stratum.
VPA-exposed ASD mice model was established as follows: VPA (Sigma-Aldrich, P4543, St. Louis, MO, USA) was dissolved in 0.9% saline to a concentration of 50 mg/mL. A single dose of VPA (500 mg/kg, i.e., 10 µL/g body weight) was intraperitoneally (i.p.) injected into pregnant mice on gestational day 12.5. Equal volume of normal saline (10 µL/g body weight) was injected as the vehicle control. For behavioral tests and Western blotting on eIF4E phosphorylation, both male and female offspring from the same litters were tested. For other tests, only male mice were used.
Antibodies
For Western blotting, the following antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA): 4E-BP1 (9452S, 1:2000), p-Thr37/46-4EBP1 (p-4EBP1, 2855, 1:2000), 4E-BP2 (2845S, 1:1000), eIF4E (9742, 1:1000), p-Ser209-eIF4E (p-eIF4E, 9741S, 1:1000), ERK1/2 (4695S, 1:1000), p-Thr202/Tyr204-ERK (p-ERK, 4370, 1:1000), MNK1(2195S, 1:2000), p-Thr197/202-MNK (p-MNK1, 2111S, 1:2000), mTOR (2972S, 1:1000), p-Ser2448-mTOR (p-mTOR, 2971S, 1:2000), p70S6K (9209S, 1:1000), p-Thr389-p70S6K (p-70S6K, 9205S, 1:1000), RPS6 (2217S, 1:1000), p-Ser235/236-RPS6 (p-RPS6, 2211S, 1:1000), HSP90 (4877, 1:1000). Antibodies against α-tubulin (A01080, 1:10000) and β-actin (A01010, 1:10000) were from Abbkine Scientific (Atlanta, GA, USA), puromycin (MABE343, 1:2000) from Millipore Corporation (Billerica, MA, USA), PSD95(ab2723, 1:1000) from Abcam (Cambridge, UK), Synaptophysin (S5768, 1:1000) from Sigma-Aldrich, and GAPDH (20035, 1:10000) from ProMab Biotechnologies (Richmond, CA, USA).
The antibodies used for immunofluorescence were as follows: p-eIF4E (NBP3-21524, 1:150) from Novus Biologicals (Centennial, CO, USA), microtubule-associated protein 2 (MAP2; ab5392, 1:200) and PSD95 (ab2723, 1:200) from Abcam, and puromycin (MABE343, 1:200) from Millipore. The secondary antibodies Alexa Fluor 488 goat anti-mouse IgG antibody (A11029, 1:200) and Alexa Fluor 546 goat anti-Rabbit IgG antibody (A11035, 1:200) were from Invitrogen (Thermo Fisher Scientific Inc, Carlsbad, CA, USA).
Behavioral tests
Social discrimination test was performed at juvenile stage (4–5 weeks old). Three-chamber test, self-grooming, and Open-field test were performed in male and female offspring at both 5 and 9 weeks old. Cognitive function tests and elevated Zero-maze test were performed only in male offspring in adulthood (9 weeks old). Mice were transferred to the behavior testing room to acclimate the environment 1 h before each experiment. In both the social discrimination test and Three-chamber test, all the unfamiliar mice were age- and sex-matched with the testing mice. The ambient illumination was 100–120 lux for the Open field test, and 40–60 lux for all other behavioral tests. All behavioral tests were recorded by an overhead video camera and analyzed by TopScan 3.0 (Clever Sys Inc, Washington, D.C., USA) [76, 77]. The experimenter was blinded to the groups of experiments.
Social discrimination test
Social discrimination test was conducted in a 40 cm (length) × 40 cm (width) × 40 cm (height) rectangular box. In the first phase, an empty wire cup was placed on one side of the arena, and the test mouse was allowed to freely explore the area for 10 min. In the second phase, a stranger mouse was placed in the cup, and the test mouse was allowed to explore freely again for 10 min. The duration of interaction with the empty cup or the stranger mouse were analyzed. The interaction index was calculated as the ratio of interaction time with the stranger mouse to interaction time with the empty cup.
Open field test and self-grooming analysis
The test mouse was put in the center of a 40 cm (length) × 40 cm (width) × 40 cm (height) arena and allowed to explore freely for 30 min [76, 77]. The travel distance and duration in the peripheral and center regions of the mice were analyzed. Additionally, the duration of grooming time of each mouse within the last 20 min of the 30-min test was analyzed. Grooming behaviors including washing face, licking paws, scratching or rubbing head and ears were counted.
Three-chamber test
Three-chamber test was carried out in a 60 cm (length) × 40 cm (width) × 20 cm (height) box with three chambers partitioned by two walls, each has a passage (5 × 5 cm) at the bottom [76, 77]. Following a 10-min habituation in the three empty chambers, the test mouse was subject to a 10-min social approach phase, in which it was allowed to explore an empty wire cup placed in one side chamber and a stranger mouse-containing wire cup in the other side chamber. The test was concluded with a 10-min social novelty phase, in which the test mouse was allowed to explore a familiar mouse-containing cup or an unfamiliar mouse-containing cup. Travel distance, the exploration time in different chambers, and sniffing time for the stranger mouse or empty cup were analyzed.
Cell culture, plasmids construction, and transfection
The Neuro-2a and HEK293T cell lines, obtained from the American Type Culture Collection (ATCC), were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, C11995500BT) supplemented with 10% Fetal Bovine Serum (Yeasen, 40130ES76) and 1% Penicillin-Streptomycin (Gibco, 15140122), at 37 °C in 5% CO2. The plasmids pCDH-3×FLAG-WT-eIF4E, pCDH-3×FLAG-eIF4E S209A (non-phosphorylatable), and pCDH-3×FLAG-eIF4E S209D (phosphomimetic) were constructed using standard cloning and mutagenesis method. Different plasmids were transfected in cells with 80% confluency using Lipofectamine LTX (Invitrogen, 15338100) following the manufacturer’s instruction.
De novo protein synthesis measurements
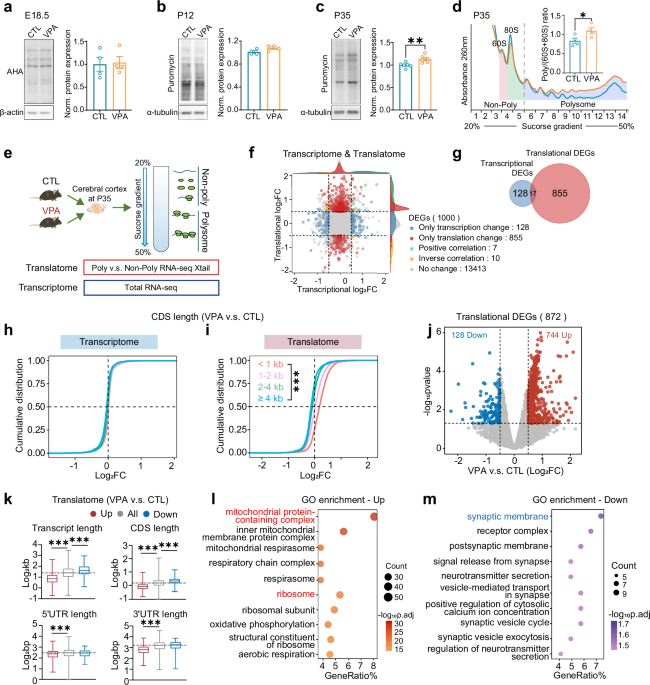
For cortical protein synthesis measurement at E18.5, BONCAT was performed by i.p. injection of 50 μg/g AHA (Click Chemistry, 1066-100), a bioorthogonal analogue of methionine, to the pregnant mice at E17.5. After 16 h, embryos were harvested and the AHA-incorporated nascent proteins were coupled with disulfide biotin-DBCO (Click Chemistry Tools, A112) by copper free catalytic azide alkyne cycloaddition reaction. Streptavidin-Dynabeads (Thermo Fisher, 11206D) were used to pulldown the biotin-coupled nascent proteins, which was measured by Western blotting using HRP-conjugated streptavidin (Thermo Scientific, N100).
For measurement at postnatal stages, mice at P12 and P35 were sacrificed, and the brains were rapidly perfused to prepare slices as described previously [77]. The brain slices (300 μm) were placed in artificial cerebrospinal fluid (ACSF; 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, and 10 mM glucose) and maintained at 32 °C for 40 min, bubbled with 95% O2 and 5% CO2. The slices were then transferred to bubbled ACSF containing 10 μg/ml puromycin (Sigma-Aldrich, P8833) for 25 min. Following the incubation period, the brain slices underwent two washes with ACSF, and the cortical and hippocampal regions were carefully separated for Western blotting using anti-puromycin antibody.
For puromycylation in cell cultures, Neuro-2a cells were transfected with plasmid for 24 h. Then puromycin (1 μM final concentration; Sigma-Aldrich, P8833) was added in the culture medium and incubated with the cells for 30 min. Cell lysates were then collected for Western blotting using anti-puromycin antibody. For in situ visualization of protein synthesis, AHA-based FUNCAT was used to visualize protein synthesis in the cytoplasm and mitochondria [69, 78]. Cells were incubated in a methionine-free medium, i.e., Dulbecco’s modified eagle medium without L-glutamine, L-methionine and L-cystine (Gibco, 21013024) supplemented with 200 μM L-cystine (MCE, HY-N0394) and 2 mM GlutaMAX (Gibco, 35050061), for 20 min to exhaust L-methionine taken up in the cells. Cells were then treated with 50 μM AHA (Invitrogen, C10102) for 30 min to label nascent proteins. To differentiate cytoplasmic and mitochondrial protein synthesis, 100 μg/mL of cycloheximide (Sigma, C7698) or both cycloheximide and 150 µg/mL chloramphenicol (MCE, HY-B0239) was pretreated the cells for 20 min before AHA was added. The cells were fixed, permeabilized, and AHA-labeled nascent proteins were covalently linked with Alexa Fluor 546 alkyne (Thermo Fisher, A10275) through a copper-catalyzed azide-alkyne cycloaddition (CuAAC) click reaction using the Click-iT Cell Reaction Buffer Kit (Invitrogen, C10269). FUNCAT signals were imaged using a Zeiss LSM 800 confocal microscope (Carl Zeiss AG).
Polysome profiling
Polysome profiling of cortical tissues was performed through a continuous 20% to 50% sucrose density gradient via ultracentrifuge with SW-4Ti rotor (Beckman, Optima XPN-100), following the protocol published previously [79]. For Neuro-2a cells, samples were lysed immediately on ice in a buffer containing 15 mM Tris-HCl pH 7.5, 300 mM NaCl, 15 mM MgCl2, 1% Triton-X100 (v/v), 0.5% Na-deoxycholate (w/v), 100 U/mL RNase inhibitor (Thermo Fisher Scientific, Waltham, MA USA), 20 mM DL-Dithiothreitol (Aladdin Holdings Group Co., Ltd, Los Angeles, CA, USA), 0.1 mg/ml cycloheximide, 0.15 mg/mL chloramphenicol, and 1×protease inhibitor (TargetMol Chemicals Inc, Wellesley Hills, MA, USA). Lysates were separated through a continuous 10% to 50% sucrose density gradient via ultracentrifuge. 15 gradient fractions from light to heavy sedimentation were sequentially collected by a Piston Gradient Fractionator (BioComp Instruments, Nova Scotia, Canada) while RNA was detected at UV absorbance 260 nm. The peak areas of different ribosome components were quantitatively calculated by OriginPro 9.0 software (OriginLab Co., Northampton, US).
RNA sequencing and translatome analysis
The samples obtained by polysome profiling were divided into non-poly fractions, containing non-translated or weakly translated mRNAs, and poly fractions, containing actively translated mRNAs. Total RNAs from non-poly and poly fractions, as well as from total homogenates, were extracted using TRIzol reagent (Invitrogen, 15596026). RNA-sequencing was performed on the DNBSEQ-T7 sequencing platform, yielding approximately 6 G of raw data per sample which were subjected to quality control using the FastQC package. The NanoTrans analysis pipeline [80] was utilized to align the data with the GRCm39 mouse genome reference (NCBI Annotation Release 109; https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001635.27/). For the translatome analysis, DEGs were identified using Xtail R package [47] based on the values calculated from the read counts of polysome mRNA over non-poly mRNA fractions. For transcriptome analysis, DEGs were identified from total RNA-seq data using DESeq2 R package [81]. DEGs were defined by |Log2FC| > 0.5 and p-value < 0.05. Volcano plots of DEGs were generated using the ggplot2 R package, and enrichment analysis for all DEGs was performed using the ClusterProfiler R package, referencing the Gene Ontology (GO) database. GSEA analysis using the ClusterProfiler R package, referencing the MSigDB GSEA database. The R version 4.2.3 was used in this study.
SFARI gene classification
Genes associated ASD were curated from the SFARI GENE database (https://gene.sfari.org/, updated on July 7, 2025) using the following criteria: Genes with ≥ 10 supporting reports (n = 472) and genes reported to be syndromic (n = 464), filtered out if there is the term “syndromic” in the description of genetic-category. Gene lists of these two classifications are provided in Table S5 and S6.
RNA extraction and quantitative real-time PCR (RT-qPCR)
RT-qPCR was conducted following the methods described previously [79]. cDNA synthesis from mRNAs in non-poly, poly, and total fractions was carried out using Moloney murine leukemia virus reverse transcriptase (MMLV-RT, Promega, M170A). qPCR amplification was performed using iQSYBR® Green Supermix (Bio-Rad, B21202) on a qPCR real-time system (Roche Light Cycler 480 Instrument). Primers for each gene were designed to amplify all possible spliced and edited variants, with the sequences listed as follows (from 5’ to 3’):
mt-Nd1-F: GGCCCATTCGCGTTATTCTT
mt-Nd1-R: AGCGTGGATAAGATGCTCGG
mt-Nd5-F: AACCAGCATTCCAGTCCTCAC
mt-Nd5-R: GTTGAGGTGGATTTTGGGATGG
mt-Co1-F: CTACCCACCTCTAGCCGGAA
mt-Co1-R: TGTGTTTAGGTTGCGGTCTGT
mt-Co2-F: GCTCTCCCCTCTCTACGCAT
mt-Co2-R: AGCAGTCGTAGTTCACCAGG
mt-Cytb-F: CCTTGACCCGATTCTTCGCT
mt-Cytb-R: GGGTGGGGTGTTTAGTGGAT
mt-Atp6-F: CGCCTAATCAACAACCGTCTC
mt-Atp6-R: CCAGCTCATAGTGGAATGGCT
mt-Atp8-F: GCCACAACTAGATACATCAACATGA
mt-Atp8-R: TGTTGGGGTAATGAATGAGGCA
Fis1-F: GAGGGCTGTTGCAGACTGAG
Fis1-R: CCAGTCCAATGAGTCCAGCC
Timm13-F: GGCTTCGGCTCGGATTTTG
Timm13-R: CACTTGTCCGTCATTCTCTGG
Tomm6-F: AGGTTCCAGACAACGTGGGA
Tomm6-R: AATGTCACTCAAGTTCCTGGC
Ndufv3-F: ATGTCTGTGAACGGAGCGG
Ndufv3-R: AGCACACTTTGTGTCTTGGGA
Ndufaf8-F: ATGTCTGTGAACGGAGCGC
Ndufaf8-R: GCGAACGCAGAGGTCCTTAC
Rps12-F: GCCAAAGCCTTAGACAAGCG
Rps12-R: GGCCTACCCATTCCCCTAGT
Rps21-F: GCCGAGGTTGATAGGACCAC
Rps21-R: ATCAGCCTTAGCCAATCGGAG
Rps29-F: GTCTGAAGGCAAGATGGGTCA
Rps29-R: TGGTAGTAGTCAGTCGAATCCA
Rpl3l-F: GAAGGGCCGGGGTGTTAAAG
Rpl3l-R: AGCTCTGTACGGTGGTGGTAA
Rpl8-F: AAGGCGCGGGTTCTGTTTT
Rpl8-R: GCTCTGTCCGCTTCTTGAATC
Rplp1-F: CAAGGCTCTGGCCAATGTCA
Rplp1-R: AGTCAAAAAGACCGAAGCCCA
Mrps26-F: CATGACCCGCCTGCCAAAT
Mrps26-R: TCTCCCGGTACTGTCGGTAG
Mrpl28-F: CCTCACGGGACCAAGTTCAA
Mrpl28-R: CCCTCTTGGAGAGCTTGTCG
LC-MS/MS and data analysis
Cerebral cortices were homogenized in ice-cold lysis buffer containing 320 mM sucrose, 5 mM Tris-HCl (pH 7.4), 1 mM MgCl2, 1 mM DTT, and protease and phosphatase inhibitor cocktail (TargeMol, C0004). The homogenate was centrifuged at 1000×g for 10 min at 4 °C to remove debris and nuclei. The supernatant was then centrifuged at 13000×g for 15 min at 4 °C, and the pellet (crude synaptosome fraction) was suspended in a solution containing 320 mM sucrose, 5 mM Tris-HCl (pH 7.4), and protease and phosphatase inhibitors. Proteins were quantified using BCA assay kit (Thermo Fisher, 23252). 80 μg of protein per sample was processed on the PreOn platform using the iST Protein Preparation Kit (PreOmics, P.O.00027, Germany), with trypsin digestion for 3 h. Digested peptides were desalted, vacuum-dried, and reconstituted in 0.1% formic acid for quantification. A total of 300 ng of peptides was loaded onto a nanoElute2 system coupled to a timsTOF HT (Bruker Corporation, USA) via CaptiveSpray ionization. Peptides were separated using a 55-min linear gradient from 2% to 37% acetonitrile (in 0.1% formic acid). Data were acquired in diaPASEF mode (m/z 300–1,500). Database searches were performed in PEAKS Studio 12 against the SwissProt Mus musculus database (17,214 entries). Parameters included trypsin specificity, up to 2 missed cleavages, peptide length 7–30 residues, variable modifications (methionine oxidation, N-terminal acetylation), and fixed carbamidomethylation. Match-between-runs, label-free quantification, and total ion current normalization were applied. Proteins with at least one unique peptide detected were identified and quantified. Differentially expressed proteins were defined as up-regulated (fold change > 1.1, p < 0.05) or down-regulated (fold change < 0.9, p < 0.05).
ATP assay
ATP levels were measured using an ATP Assay Kit (Beyotime, S0026). Cortical tissues were immediately homogenized in the ATP detection lysis buffer, centrifuged at 4 °C at 12000×g for 5 min, and the supernatant was collected for the measurement of ATP content using Infinite F500 (TECAN, Switzerland).
Transmission electron microscopy (TEM) and image processing
Mice were euthanized and the cerebral cortices were rapidly dissected on ice. The mPFC was micro-dissected out and immediately fixed in the TEM fixative solution for 2 h at room temperature before placed at 4 °C. Subsequent steps for routine TEM, including dehydration, sectioning, and imaging, were carried out. Sections were imaged using a Hitachi TEM system at 80.0 kV. Analyses of the number and morphology of mitochondria within the synaptic areas, the number of presynaptic vesicles, and the morphology of synapses were performed using ImageJ software (National Institutes of Health, Bethesda, MD, United States).
Mitochondrial complex activity assays
Different mitochondrial complex activity levels were measured using the following kits correspondingly: Mitochondrial Complex I Activity Assay Kit (Elabscience, E-BCK149-M), Mitochondrial Complex II Activity Assay Kit (Elabscience, E-BCK150-M), Mitochondrial Complex III Activity Assay Kit (Elabscience, E-BCK151-M), Mitochondrial Complex IV Activity Assay Kit (Elabscience, E-BCK152-M), and Mitochondrial Complex V Activity Assay Kit (Elabscience, E-BCK153-M). Briefly, cortical tissue was homogenized and centrifuged at 600×g for 5 min at 4 °C, and the supernatant was then centrifuged at 11000×g for 10 min at 4 °C. The pellet was resuspended and sonicated for 1 min at 4 °C, and centrifuged at 11000×g for 10 min at 4 °C. The supernatant was collected for the measurement of different mitochondrial complex activities.
Primary cortical neuron culture from VPA-exposed embryos
A single dose of VPA (500 mg/kg body weight) was i.p. injected in pregnant C57BL/6 mice on gestational day 12.5, and normal saline was injected as the vehicle control. Primary cortical neurons were cultured from VPA- or saline-exposed embryos at E17.5 following a protocol published previously [82, 83]. Briefly, the cortical tissues were trypsinized to obtain a single-cell suspension, and then seeded on 18-mm diameter coverslips coated with 1 mg/mL poly-D-lysine (Millipore, A-003-E) at a density of 2.5×104 cells per coverslip, and cultured in Neurobasal Medium (Gibco, 21103049) supplemented with 2% B27 (Gibco, 17504044), 1 mM L-Glutamine (Gibco, 25030081), and 0.18% D-Glucose (Gibco, A2494001). Half of the medium was changed every three to four days.
Mitochondrial membrane potential assay
Mitochondrial membrane potential in primary cortical neurons was measured using the Mitochondrial Membrane Potential Assay Kit with TMRM (Beyotime, C2001S). Briefly, TMRM was diluted to 1×solution and added to primary cortical neurons at 15 days in vitro (DIV) for 30 min at 37 °C. Carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a mitochondrial membrane potential depolarization agent, was added to cells at a concentration of 10 μM for 20 min pre-incubation, acting as a negative control. After incubation, the cells were washed twice and TMRM fluorescence was photographed in live cells using Zeiss LSM 800 confocal microscope.
Immunofluorescence
To analyze dendritic PSD95 puncta, primary cortical neurons at 14 DIV were fixed and immunostained with mouse anti-PSD95 and rabbit anti-MAP2 antibodies as previously described [84]. Images were acquired using Zeiss LSM 800 confocal microscope with a 63× objective (Carl Zeiss AG). PSD95 puncta density was quantified in MAP2-positive dendrites using ImageJ software.
To analyze the levels of p-eIF4E and puromycin in the cortex, cryosections (20 μm) were made using a frozen microtome (Leica, CM1950). The sections were permeabilized with 0.3% Triton X-100 in PBS for 30 min, then blocked with 3% bovine serum albumin (Solarbio, A8020) and 10% goat serum (GENOM, GNMGS100) at 37 °C for 1 h. The sections were then incubated with rabbit anti-p-eIF4E and mouse anti-puromycin antibodies at 4 °C overnight, followed by 1 h incubation at room temperature. Secondary antibodies Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 546 goat anti-rabbit IgG were applied at 37 °C for 1 h. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, D9542, 1:1000) for 10 min. Sections were mounted with Antifade Mounting Medium (Beyotime, P0128S) and imaged on Zeiss LSM 800 confocal microscope.
Mitochondrial respiratory chain enzyme analysis
The Oroboros O2k experiment was conducted following an established protocol with the Oroboros O2k platform (Oroboros Instruments, Austria) [85]. HEK293T cells were plated in 6-well plates and detached by treating with a 0.05% trypsin-EDTA solution (Gibco, 15400054) for 30 s. Afterward, the cells were counted and adjusted to a density of 1×106 cells per 100 µL. Only two samples were tested simultaneously. Basal respiration was recorded after 10-min equilibration at 37 °C. Oligomycin (5 nM final concentration; Sigma-Aldrich, O4876) was then added to inhibit ATP synthase (Complex V). Maximal respiration was induced by titrating carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 1 µM final concentration; Sigma-Aldrich, C2920). Rotenone (0.5 µM final concentration; Sigma-Aldrich, R8875) was injected to inhibit Complex I. Antimycin A (2.5 µM final concentration; Sigma-Aldrich, A8674) was added to inhibit Complex III. Oxygen consumption rates were normalized to cell count and expressed as pmol O₂/s/10⁶ cells.
Drug preparation and treatment
eFT508 (Tomivosertib; MedChemExpress LLC, Monmouth Junction, NJ, USA) was dissolved in DMSO at a concentration of 5 mg/mL as a stock solution. The working drug solution was made by freshly diluted the stock solution in normal saline with PEG300 (40% v/v) and Tween-80 (5% v/v) to a concentration of 1 mg/mL. The prenatally VPA-exposed male mice were randomly allocated to vehicle group (VPA-DMSO) and drug-treated group (VPA-eFT508), and mouse in each group received 4 times daily i.p injections of vehicle or eFT508 (1 mg/mL) correspondingly at postnatal week 3. Behavior tests including Open field test, self-grooming analysis, Three-chamber test, and elevated Zero-maze test were performed at both postnatal Weeks 4–6 and Week 9. A single drug dose was administered 1 h prior to each behavioral experiment and tissue collection.
Statistical analysis
No a priori power analysis was conducted because the sample sizes for each experimental group were based on pilot data and published studies [76, 77, 79]. All experiments were conducted in a blinded manner. Statistical analyses were carried out using GraphPad Prism 9.2.0. The results were presented as mean ± SEM. Distribution normality (Shapiro–Wilk test) and homogeneity of variance of the data were checked before parametric analyses. No exclusion criteria were pre-determined in our tests. Differences between the two groups were assessed using two-tailed t-test, and multiple-group comparisons were analyzed using one-way analysis of variance (ANOVA) followed by post hoc Tukey’s multiple comparisons. Differences between distributions were compared using KS test or two-sample z test. P-value less than 0.05 was considered statistically significant for all statistical analyses. Detailed statistical information of each experiment was provided in Table S10.